density n(r):
Eks [n(r)] = Tni [n(r)] + n(r) vext (r)dr
1 n(r)n(r′ )
+ drdr + Exc [n(r)]
2 |r − r′|
whereTni [n(r)] represents the non-interacting particles kinetic
energy, vext (r) the interaction potential of the ions with the
International Journal of Scientific & Engineering Research Volume 4, Issue 1, January-2013 1
ISSN 2229-5518
First Principle Band Structure Calculations of
Zinc-Blende BN and GaN Compounds
1 Nawzad A. Abdulkareem, 2 Badal H. Elias
and GW approxim ation (in conjunction with the ABINIT package) is used to investigate latt ice const ant paramet er (a) and band structure for zinc-
blende BN and GaN. To be more specific, the Perdew-Wang92 (PW 92) and Perdew-Burke-Ernzerhof (PBE) flavors have been employed for
exchange-correlation term of LDA and GGA, respectively. Lattice const ant (a), band gap energy (Eg ) have been calculat ed using LDA and GGA approxim ations. The GGA results for (a) are strongly agree with the experimental and much more accurat e than LDA values. The values of a in both LDA and GGA m ethods are well accurat e when compared with the other theoretical works. The Eg values obtained are in a good agreem ent with other theoretical works especially for ZB-BN. On the other hand, the Eg values are not in good agreem ent with the experimental due to the well known band gap problem of density functional theory (DFT). To improve the Eg value, GW approximation has been used. It was found that the
improvem ent is better for ZB-BN than ZB-GaN.
—————————— ——————————
HE III-V semiconductors consist of two elements, one is from group III and another is from group V in the periodic table.
III-V semiconductors are more important in optoelectronics because of their wide band gap. They have applications in specific areas such as wireless communications. III-Nitrides (III-N) form a specific subgroup of the III-V compounds with basic crystal structures, Hexagonal wurtzite (WT) structure and cubic zinc- blende structure (ZB). III-N semiconductors are characterized by high iconicity, very short bond length, low compressibility and high thermal conductivity [1, 2 and 3]. These properties make the III-N semiconductors interesting and very useful. These materials can therefore be used for short wavelength light-emitting diodes (LED), laser diodes [4], and optical detectors, as well as for high-
temperature, high power, and high frequency devices [5 and
coating materials [4].
The calculations were performed using Pseudopotential-Plane wave method within the framework of the density functional theory (DFT) [8 & 9]. The pseudopotentials for all atoms were generated according to the scheme of Troullier and Martins [10].In the density functional formalism, the total electronic energy of the ground state is given as a functional of the total electron charge
density n(r):
Eks [n(r)] = Tni [n(r)] + n(r) vext (r)dr![]()
![]()
1 n(r)n(r′ )
+ drdr + Exc [n(r)]
2 |r − r′|
whereTni [n(r)] represents the non-interacting particles kinetic
energy, vext (r) the interaction potential of the ions with the
1 n (r)n(r ) ′
![]()
6].Among III-N semiconductors, the Boron nitride (BN) and
electrons (external potential), ∬
2
drd r
lr-r′ l
the electrostatic
Gallium nitride (GaN) with indirect and direct band gaps have attracted extensive experimental and theoretical interest. BN and GaN, like most III-V compounds, have WZ and ZB structures. The main focus of this project is ZB-BN and ZB-GaN. ZB-BN is formed under high pressure, high temperature treatment of WZ- BN. It is the second hardest material known after diamond. It has high thermal conductivity and excellent wear resistance. It can be used to make cutting tools due to its hardness and to make optoelectronics devices [7].
ZB-GaN used in bright (LED),and recently, it was used as solid-state ultraviolet (UV) detectors in medical diagnostic, chemical and biological analysis, flame detection, andmissile warning, as well as for UV LEDs in food and water treatment, and
interaction among the electrons and Exc [n(r) the exchange-
correlation energy. In the present work, the exchange- correlation
energy is treated using both the local density approximation (LDA) with the Perdew and Wang92 flavor [11], and the generalized gradient approximation (GGA) with the Perdew, Burk and Ernzerhof (PBE)flavor [12].The ab initio calculations show the typical underestimate of the band gap with respect to the experimental one, due to the use of the Density Functional Theory (DFT) formalism. Therefore, the Kohn-Sham (KS) eigenvalues are usually corrected by using Hedin’s GWapproximation [13] to evaluate the quasiparticle energies instead of the considered
fictitious KS particles. The quasiparticle energy E QP can be
obtained from the so-called quasiparticle equation, which can be
written as:
2
the hardness and large bulk modulus make it ideal protective
————————— ———————
![]()
∇
− + vH
2
+ vext
ljJQP (r)
+ ∑(r, r′ ; Ei QP )ljJ
(r′)dr′ = Ei QP
ljJ
QP (r)
i i
Nawzad A. Abdulkareem, Faculty of Science, Physics Department, Zakho
University, Iraq. E-mail: nawzad_kori @yahoo.com
Badal H. Elias, Faculty of Science, Physics Department, Duhok University,
Iraq. E-mail: badel_hyder@yahoo.com
where vH and vext are Hartree potential and external potential,
QP
respectively,∑ is called self-energy, andljJi
wave function.
(r)is the quasiparticle
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 4, Issue 1, January-2013 2
ISSN 2229-5518
Boron Nitride (BN) and Gallium Nitride (GaN) in ZB structure are such that B (and Ga) atom is located at 000 coordinates while![]()
N atom is located at 1 1 1
4 4 4
coordinates. It is well known that the
atomic numbers of B, Ga and N are 5, 7 and 31, respectively. So,
the electronic configuration for B is1s22s22p1, for N is 1s2 2s22p3 and for Ga is 1s2 2s2 2p63s23p64s23d104p1. In the present work, the pseudopotentials have been used which treats B: 2s2 2p1, N: 2s22 p3 and Ga: 4s2, 4p1 as the valence states. In the present work, first,
convergence tests to ETOT were performed in order to determine the cut-off energy ( Ecut ) and the number of k-point grids (ngkp).The ETOT convergence was obtained forEcut being equal to
828.381(eV) for ZB-BN, while for ZB-GaN compound Ecut
is830.084 (eV). The other convergence test corresponds to the
sampling of the BZ. One should examine different grids for
increasing resolution. TheETOT has been converged for the 6×6×6
grid. The Symmetries are used to decrease the number of k-points
needed to sample the BZ, so that only the irreducible part of it can be sampled. The number of k-points that has been generated
automatically by the code and used in the lattice constant (a)
calculations is 28 k-points, while the number of k-points used in
LDA and GGA present calculations of band structure was set to
40. Finally, the number of k-points used in GW corrections to band gap was restricted in 4 points for ZB-BN, due to its indirect
band gap(Г1Sv ,Г1c ,X1Sv and X1c ), and 2 points for ZB-GaN (Г1Sv
and Г1c ).
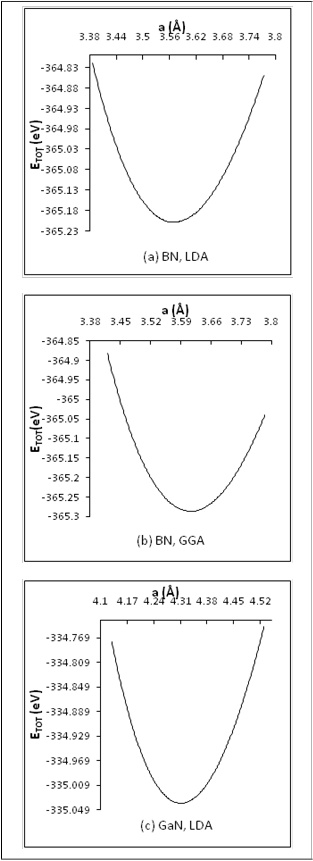
The lattice constant ( a ) of ZB-BN and ZB-GaN has been
determined using LDA and GGA by calculating the total energy
(ETOT ) for many different values of (a). The ETOT has been plotted against the corresponding (a) values (Figure 1), and the value of (a) (the equilibrium value) has been chosen, which corresponds to
the least minimum energy.
The equilibrium value of a for ZB-BN in LDA calculations
was obtained to be 3.57 (Å), while in GGA calculationsit was
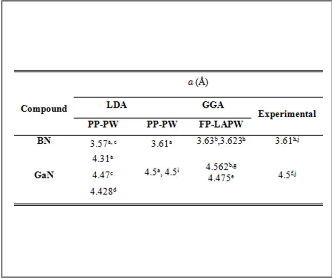
3.61(Å). The results of ( a ) is given in table (1) with the
experimental and results of other theoretical works. The present
values of a in GGA (with PP-PW) calculations for ZB-BN were
found to be exactly agreeable with the experimental value of
reference [14], while the a value in LDA (with PP-PW)
calculations is by (1.1%) smaller than the experimental value. On
the other hand, the value of a in LDA calculations has the exact
value when compared with the value of the full potential linear
augmented plane waves method (FP-LAPW) presented by the
reference [15], while a value in GGA calculations (with PP-PW) is
0.2% and 0.5% smaller than other work values of references [6
and16]whereFP-LAPW have been used. In general, the avalue in
GGA calculations has been found to be much better than avalue in
LDA calculation.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 4, Issue 1, January-2013 3
ISSN 2229-5518
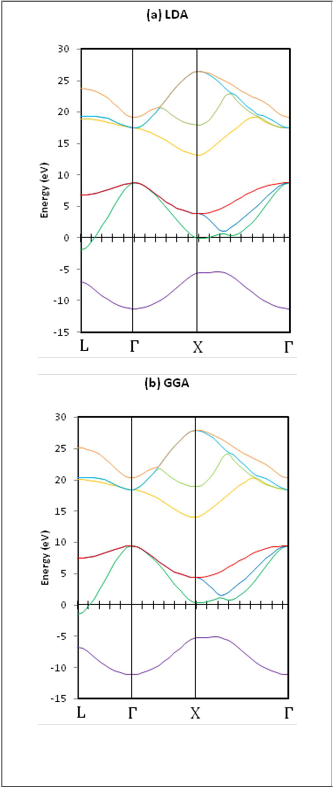
The band structures and band gap energies have been
computed for ZB-BN and ZB-GaN compounds using both
LDA and GGA calculations with and without GW
approximation employing the a values that were obtained
from GGA calculations.
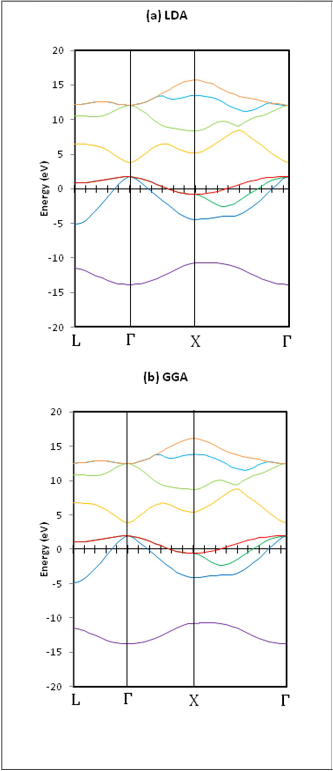
Fig. (2) shows that GGA results for band energies are higher than the LDA results, and that the difference in the conduction band are larger than the difference in the valence band energies.
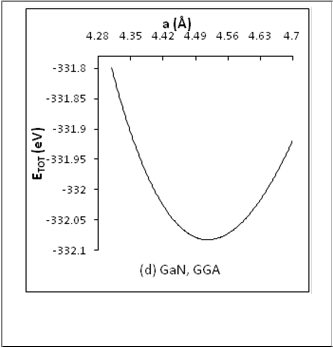
Fig.(1) The total energy ETOT plot sversus lattice constant (a) for
ZB-BN and ZB-GaN in LDA and GGA calculations.
The present value for(a) in GGA calculations for ZB-GaN
was found to be 4.5 (Å), which agrees exactlywith the
experimental and theoretical valuesevaluated by reference
[17], while a value in LDA calculationswas found to be smaller by 4.2% than the experimental value. The a value in GGA
calculation is 0.5% larger than that of reference[18] calculated
in FP-LAPW method and its value is smaller by 1.3% than the
values computed by references[16 and 19] using FP-LAPW
method. The value ofain LDAwas found to be smaller by 2.4%
and 3.5 % than other theoretical values [20 and 15]using PP-
PW, which is the same method used here. In general, the a
valuein GGA was found to be much better thanthe a
valueusing LDA.
TABLE (1)
THE PRESENTLY CALCULATED AND OTHER THEORETICAL AND EXPERIMENTAL LATTICE CONSTANTS IN Å FOR BN AND GAN IN ZB STRUCTURE.
aPresent work, bReference[16], cReference[15], dReference[20],
eReference[18], fReference[21],gReference[19], hReference[14],
iReference[17], jReference [6].
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 4, Issue 1, January-2013 4
ISSN 2229-5518
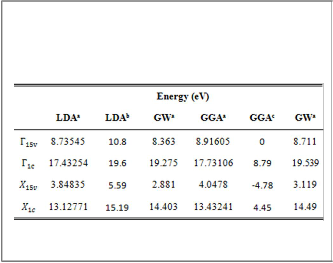
The energies computed here using LDA are in good agreement when compared with the energies obtained by the reference [15] because in both works the PP-PW method has been used, while the energies computed here using GGA are higher than the energies presented by the reference [16] where FP-LAPW method has been used.
TABLE (2)
THE ENERGIES (IN eV) AT HIGH SYMMETRY POINTS OF ZB- BN CALCULATED IN LDA AND GGA METHODS W ITH AND
WITHOUT GW APPROXIMATION EMPLOYING(A)OBTAINED
FROM GGA.
aPresentwork,bReference[15], cReference[16].
The theoretical Eg values calculated in LDA and GGA
methods with and without GW approximation with the
experimental Eg values are listed intable (3). The present work results of Eg in LDA and GGA do not agree with the
experimental value. On the other hand, there is fair agreement
when the calculated Eg values using GW approximation are compared with the experimental. The value of Eg using LDA
is smaller than Eg value given in reference [15], and the value of Eg using GGA is close to the Eg value obtained by the
references [16 and 22] where FP-LAPW method and PP-PW
method have been used, respectively.
TABLE (3)
THE EXPERIMENTAL BAND GAP VALUES (IN eV) AND
THEORETICAL BAND GAP VALUES FOR ZB-BN CALCULATED IN LDA AND GGA METHODS WITH AND W ITHOUT GW
APPROXIMATION EMPLOYING (a)VALUE OBTAINED FROM
GGA.
Fig.(2)Band structure of ZB-BNin (a) LDA calculations and (b)
GGA calculations, the (a) value obtained from GGA calculations
was employed.
aPresentwork,bReference[15],cReference[16],dReference[22]
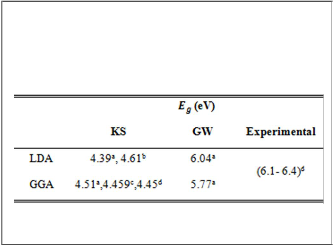
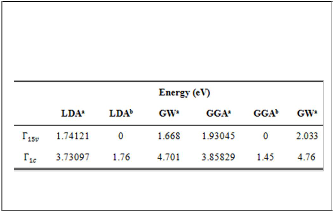
Table (2) contains the energies of the most important high
symmetry points, including Г1Sv and X1c points, calculated in
LDA and GGA calculations with and without GW
approximation for avalue obtained from GGA.
The band structure and Eg of GaN were also computed in
LDA and GGA with and without GW approximation
employing thea value obtained from GGA.The band structure
in LDA and GGA methods illustrated in the figure (3).
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 4, Issue 1, January-2013 5
ISSN 2229-5518
in Г1c , this leads to the smaller Eg for ZB-GaN in GGA than
LDA.
TABLE(4)
THE ENERGIES (IN eV) AT HIGH SYMMETRY POINTS OF ZB-
GAN CALCULATED IN LDA AND GGA METHODS W ITH AND
WITHOUT GW APPROXIMATION EMPLOYING (a)OBTAINED
FROM GGA.
aPresent work, bReference[5]
The values of the Eg for ZB-GaN in LDA and GGA
calculations with and without GW approximation of the
present work with experimental and other theoretical works
are listed in the table (5). The Eg value in LDA and GGA are
smaller than experimental by 39.7% and 41.5%, respectively,
this shows that the band gap problem of DFT. TheEg values in
LDA and GGA are in difference with the other LDA and GGA
values by (5-23) % presented by the other works. The GW
update on LDA and GGA forEg values are in fair agreement with the experimental one,the value ofEg obtained from GW
update on the LDA value has found to be 3.03 (eV) while on
the GGA has found to be 2.72 (eV) and the error with the
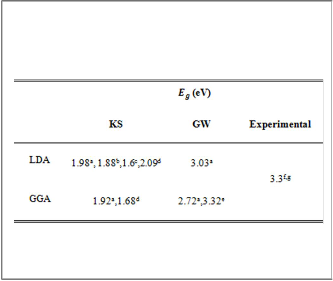
experimental reduced to be 7 % and 17%, respectively.
TABLE(4)
THE ENERGIES (IN eV) AT HIGH SYMMETRY POINTS OF ZB-
GAN CALCULATED IN LDA AND GGA METHODS W ITH AND
WITHOUT GW APPROXIMATION EMPLOYING (a)OBTAINED
FROM GGA.
Fig.(3)Band structure of ZB-GaN in (a) LDA calculations and (b)
GGA calculations, the (a) value obtained in GGA calculations was
employed.
The Г1Sv andГ1c calculated here and by other works in LDA
and GGA calculations with and without GW approximation
are listed in table (4). The Г1Sv andГ1c energies are higher than
the results obtained in the reference [5]. On the other hand, the
GGA values for Г1Sv and Г1c energies are higher than LDA
values and the difference in the Г1Sv is more than the difference
aPresent work, bReference[20], cReference[5], dReference[23],
eReference[22],fReference[24],gReference[25]
The present study concludes the following:
1- Lattice constant parameter values (a) calculated in GGA
method for both ZB-BN and ZB-GaN agree with the
experimental values and are much better than (a) values
in LDA method.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 4, Issue 1, January-2013 6
ISSN 2229-5518
2- The present calculations have shown (proved) indirect and direct band gaps for each ZB-BN and ZB-GaN, respectively.
3- Band gap energies (Eg ) in LDA and GGA calculations are
not accurate when compared with the experimental band
gap values and that is due to the band gap problem of DFT, while it is in fair agreement when they have been calculated using GW approximation with both LDA and GGA methods.
[1] N. E. Christensen and I. Gorczyca, Optical and Structural Properties of III-V Nitrides Under Pressure, Phys. Rev. B 50, 4397 (1994).
[2] B. Abbar, B. Bouhafs, H. Aourag, G. Nouet and P. Ruterana, First- Principles Calculations of Optical Properties of AlN, GaN, and InN Compounds Under Hydrostatic Pressure, Phys. Stat. sol. (b) 228, 457 (2001).
[3] I. Gorczyca and N. E. Christensen, Band Structure and High-Pressure Phase Transition in GaN, AlN, InN and BN, Physica B 185, 410 (1993).
[4] Bo-Ting Liou, Chieh-I Liu and Yen-KuangKuo, Investigation of Material Properties for ZincblendeAlGaN Alloys Applied in UV LEDs, Proc. of SPIE, 6669 (2007).
[5] C. Stampel and C. G. VAN de Walle, Density-Functional Calculations for III-V Nitrides Using the Local-Density Approximationand the Generalized Gradient Approximation, Phys. Rev. B 59, 5521 (1999).
[6] K. Kim, W. R. L. Lambrecht and B. Segall, Elastic Constants and
Related Properties of Tetrahedrally Bonded BN, AlN, GaN, and InN, Phys. Rev. B 53, 16 310 (1996).
[7] C. H. Ming, Electronic Properties of Compound Semiconductor, City University of Hong Kong, Department of Physics Materials Science (2008).
[8] P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev
136, 542 (1964).
[9] W. Kohn and L.J. Sham, Self-Consistent Equations Including
Exchange and Correlation Effects, Phys. Rev. 140, A1133 (1965).
[10] N. Troullier and J. L. Martins, Efficient Pseudopotentials for Plane- Wave Calculations, Phys. Rev. B 43, 1993 (1991).
[11] J. P. Perdew and Y. Wang, Accurate and Simple Analytic
Representation of the Electron-Gas Correlation Energy, Phys. Rev. B
46, 12947 (1992).
[12] J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized Gradient
Approximation Made Simple, Phys. Rev. Lett. 77, 3865 (1996).
[13] L. Hedin, New Method for Calculating the One-Particle Green’s
Function with Application to the Electron-Gas Problem, Phys. Rev.
139, A796 (1965).
[14] A. Zaoui and F. EI Haj Hassan, Full Potential Linear Augmented Plane Wave Calculations of Structural and Electronic Properties of BN, BP, BAs and BSb, J. phys. Condens. Matter, 13, 253 (2001).
[15] W. H. You, X. Hui, Z. NingDan and Z. PengHua, The Dielectric and
Dynamical Properties of Zinc-Blende BN, AlN and GaN From First- Principle Calculation, Sci. China Ser. G-Phys. Mech. Astron. 51, 1037 (2008).
[16] B. D. F. Zpeedeh, First Principle Electronic Structure Calculations of Ternary Alloys. BNxP1-x, GaxB1-xN and BxIn1-xN in Zinc Blende Structure, Master Thesis, An-Najah National University, Nablus- Palestine (2008).
[17] M. Fuchs, J. L. F. Da Silva, C. Stampfl, J. Neugebauer, and M.
Scheffler, Cohesive Properties of Group-III Nitrides: A Comparative Study of all-Electron and Pseudopotential Calculations Using the Generalized Gradient Approximation Phys. Rev. B 65, 245212 (2002).
[18] S. Berrah, H. Abid, and A. Boukortt, The First Principle Calculation
of Electric and Optical Properties of AlN, GaN and InN Compounds Under Hydrostatic Pressure, Lashkaryov Institute of Semiconductor Physics, National Academy of Sciences of Ukraine,V. 9, 12. (2006).
[19] R. Mohammad, The Electronic Band Structure of III (IN, AL, GA)-V
(N, AS, SB) Compounds and Ternary Alloys, Middle East Technical
University (2005).
[20] M. Palummo, L. Reining, R. W. Godby, C. M. Bertoni and N.
Bornsen, Electronic Structure of Cubic GaN with Self-Energy
Corrections, Europhys. Lett, 26, 607 (1994).
[21] I. Vurgaftman, J. R. Meyer and L. R. Ram-Mohan, Band Parameters for III–V Compound Semiconductors and Their Alloys, App. Phys. Rev. 89, 5815 (2001).
[22] M. Shishkin and G. Kresse, Self-Consistent GWCalculations for
Semiconductors and Insulators, Phys. Rev. B 75, 235102 (2007).
[23] B. Daoudi, M. Sehil, A. Boukraa and H. Abid, FP-LAPW Calculations of Ground State Properties for AlN, GaN and InN Compounds, Int. J. Nanoelectronics and Materials, 65 (2008).
[24] D. Vogal, P. Kruger and J. Pollmann, Structural and Electronic
Properties of Group III-Nitrides, Phys. Rev. B 55, 12, 836 (1997).
[25] M. Rohlfing, P. Kruger and J. Pollmann, Role of SemicoredElectrons
in Quasiparticle Band-Structure Calculations, Phys. Rev. B 57, 6485
(1998).
IJSER © 2013 http://www.ijser.org