International Journal of Scientific & Engineering Research, Volume 2, Issue 1, January-2011 1
ISSN 2229-5518
Damage-Based Theories of Aging and Future
Treatment Schemes
Angus Hung
Abstract— Organismal senescence has been postulated to result from a myriad of biological factors, ranging from environmental facets to detrimental internal mechanisms. However, a specific cause of aging is currently unknown. Two branches of aging theories have been proposed: damage-based theories and programmed theories. Here we examine the numerous kinds of promulgated damage-based theories. The possible therapies for combating aging in humans that may be employed in the near future will also be discussed.
Index Terms—aging; lifespan; longevity; senescence; damage-based theories of aging
—————————— • ——————————
GING is a universal phenomenon present among all known mammalian species. As an organism is sub- jected to biological aging after reaching sexual ma- turity, it gradually loses cellular and biological function: organs become impaired, cancerous cells develop, fertility is lost, and the organism inevitably becomes more prone to age-related diseases. These degrading attributes of bio- logical aging reach a climactic end when the organism experiences the ultimate consequence of aging: death. For humans, aging is not only fatal, but also costly. The elder- ly must constantly undergo treatements for the age- associated diseases which inevitably follow with the onset of aging. They pay thousands of dollars for medicines, surgeries, and healthcares simply to mitigate the effects of aging; such a disposition of finances is futile, for it merely postpones the death which will eventually be caused by aging. Ostensibly, aging is an undesirable process and
must be cured.
Luckily, aging research has greatly accelerated over
the years. Research organizations such as Aubrey de
Grey’s Strategies for Engineered Negligible Senescence Foundation have been specifically established with the single intention of curing aging. Studies of aging have strived to answer the fundamental inquiry of biogeron-
tology: what causes aging? In the past century, theories that have been formulated to answer this question fall into two categories: damage-based theories (commonly called “wear and tear” theories) or programmed theories of aging. The general consensus for damage-based theo- ries is that organisms accumulate detrimental toxins and damage at the molecular level throughout their lives, leading to the onset of aging. On the other hand, the gen- eral idea of programmed theories of aging is that aging is an intrinsic process programmed by an organism’s own genetic code. Both classes of these aging theories are ri- veting and have been supported by much scientific re- search. However, each of these biological senescence theories has faults: damage-based theories are unable to explain how similar animals have dramatically different
lifespans and programmed-based theories are unable to explicate how natural selection would promote such a detrimental process in the animal’s genome. Several types of damage-based theories of aging will be explicitly ex- amined throughout this paper.
First proposed by Denham Harman in the 1950s, the free radical theory of aging states that organismal senescence occur as a result of accumulated free radicals over a given period of time. Free radicals are atoms or molecules with an unpaired electron in their outershell: such is the cha- racteristic which makes numerous free radicals highly reactive. Reactive oxygen species (ROS), a type of free radicals, are characterized by their possession of oxygen and high chemical reactivity. When ROS levels are in- creased astronomically, cell structures degrade and and oxidative stress occurs. Essentially, a discrepancy be- tween the production and detoxification of ROS leads to oxidative stress, and thus damage. Human age-associated diseases like atherosclerosis, Parkinson’s disease, and Alzheimer’s disease are all characterized by oxidative stress. Figure 1 exemplifies the correlation between ROS and age-related diseases. Due to the correspondence be- tween aggregated ROS levels and aging-associated dis- orders, free radicals seem to be an extremely pertinent factor in the aging process.![]()
Fig. 1 – Because of the high reacvity of ROS, increasing ROS
levels will lead to damage and age-related diseases.
The accumulative and pernicious free radicals and
ROS can be derived from a plethora of metabolic
IJSER © 2010 http://www.ijser.org
2 International Journal of Scientific & Engineering Research, Volume 2, Issue 1, January-2011
ISSN 2229-5518
processes throughout the human body, most noticeably the mitochondrion. During oxidative phosphorylation, energy is taken from those derived from the oxidation of glucose (among other foods) to create the energy mole- cule necessary for a cell’s function: adenosine triphos- phate. Throughout this process, some reactive oxygen molecules are spontaneously released from the mito- chondrion. Another kind of reactive oxygen species, anion superoxides, are created through the reduction reaction of triplet-state oxygen. The reduction is catalyzed by enzymes such as NADP oxidases; they can also be mediated by nonenzymatic activity through redox reac- tive compounds of the mitochondria’s electron transport chain. Evidently, metabolic processes, especially those occurring near the mitochondrion, signify an important role in biological senescence through their production of free radicals and reactive oxygen species.
To combat the detrimental products of cellular meta- bolism, particukar enzymes are employed to mitigate the havoc caused by ROS. Enzymes such as catalase and su- peroxide dismutase lessen the damage of ROS by simply converting them to the products of oxygen and water through an enzymatic reaction. However, the conversion of metabolic products is not perfect: some ROS molecules are often left behind, leading to a gradual accumulation of deleterious substances throughout cells. Indeed, the ef- fects of ROS and other free radicals are felt as organisms age, leading to age-associated diseases such as Alzhei- mer’s disease. Aggregated ROS and free radicals steadily degrade cellular macromolecules, including deoxyribo- nucleic acid. The implications of genetic degradation are discussed next.
The DNA damage theory of aging states that accumu- lated damage to deoxyribonucleic acid is the factor that leads to the onset of biological aging. The DNA damage theory applies mainly to nuclear DNA, although recent studies have shown that mutations in mitochondrial DNA also function in limiting an organism’s lifespan. Damage to DNA is caused either by internal chemical reactions or external sources which interfere with the natural process of DNA replication. The two types of damage pertinent to the DNA damage theory include physical DNA damage and base-pair mutations. The physical deformities classified as damage include double or single strand breaks in the nucleic acid molecules. These abnormalities are easily recognized by enzymes which can then proceed to repair the physical damage. The repair of physical DNA damage is usually feasible as long as extra genetic information, whether from homo- logous chromosomes or the undamaged complementary DNA strand, is available. Antithesis to this classification of DNA damage is the creation of mutations, which are essentially modifications in the base-pair sequence of
DNA. Unlike physical DNA damage, base-pair mutations are impossible to repair. A known biological mechanism employed by cells to prevent the onset of mutations is through DNA polymerase’s DNA proofreading. Al- though strong DNA proofreading mechanisms are present in DNA replication of the nucleus, mutations still occur, with an average of one mutation per 108 replicative generations. Mitochondrial mechanisms of proofreading mitochondrial DNA also have been observed, including exonuclease proofreading by mitochondrial DNA poly- merase. However, higher rates of mutation have been observed in the mitochondrion than in the nucleus, sug- gesting the evolutionary relics of mitochondria and their primitive tactics of DNA replication. Nonetheless, muta- tions accumulate both in the nuclear and mitochondrial DNA. DNA mutations, in addition to the physical wreck created by internal and external sources, all hasten atro- phication of deoxyribonucleic acid.
The hypothesis of a distinct relationship between ge-
nome mutations and aging is highly supported because of
the age-related mutations that accumulate throughout the
cell as time passes. Indeed, genome mutation occurs in all
human tissues, most noticeably in the brain, kidney, liver,
and muscle. These cellular mutations have been linked to
a decline in gene expression. As age-associated mutations
accumulate, decline in gene expression is observed, lead-
ing to the symptons related to aging.
More support for the DNA damage theory of aging lies
in premature aging disorders. One rare aging disease,
Hutchinson–Gilford progeria syndrome (or simply pro- geria), has been known to result in premature aging. In- dividuals suffering from progeria are characterized by age-associated symptoms at an extremely young age.
Cardiovascular problems, wrinkles, and fragile bones are all symptoms of progeria. The accelerated aging disease has been linked to its defect of nuclear lamin A. Nuclear lamin A is a protein located within the interior edge of the nucleus, and functions in the organization of processes such as the synthesis of DNA and RNA. Progeria results in the unavailability of lamin A in the individual. The lack of this compulsory protein is associated with the age- related symptoms at a young, which is presumably due to damage to the nucleus and the molecules within it, in- cluding DNA.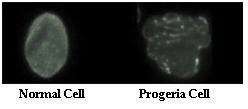
Fig. 2 – A normal cell is shown here in juxtaposition with a progeria cell. Progeria cells lack lamin A, leading to their structural deformity.
IJSER © 2010 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 2, Issue 1, January-2011 3
ISSN 2229-5518
As seen in figure 2, progeria cells accumulate damage at a faster rate than normal human cells. The reason for such a cellular deformity is presumably because of the lack of lamin A, a compulsory cellular protein. Without it, the nucleus and its components are damaged, including DNA. Thus, the inherited genetic accelerated aging dis- ease is an existing manifestation of the implications brought about by the DNA damage theory of aging.
The telomere theory of aging simply states that the suc- cessive shortening of telomeres results in the onset of ag- ing. Telomeres are the structural ends of chromosomes characterized by noncoding repetitive sequences of DNA. Presumably, telomeres protect the ends of the chromo- some from DNA degradation.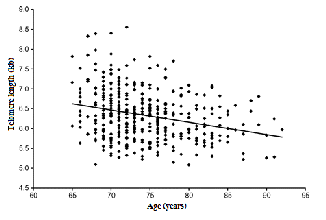
During cell division, the chromosomal ends of chromo- somes shorten successively. Such a curtailing in DNA is ex- plained by the imperfection of DNA replication itself. This end replication problem in DNA replication exists in euka- ryotes only, due to their linear DNA. Therefore, telomeres shield DNA from successive shortening in cell division. However, telomeres only postpone the cellular conundrum of DNA shortening. Eventually, cells reach a point where they can no longer divide, known was Hayflick’s limit. The halt in cell division is an obvious sign of cellular senescence, which is presumably to result from the shortening of telo- meres. Essentially, successive mitotic division results in the shortening of telomeres, resulting in cellular senescence. Cellular senescence then leads to the onset of aging (since cells are the basic units of organisms it would be logical to deduce that organisms as a whole age if cells alone do). The relationship between aging and telomeres is depicted in fig- ure 3.
Fig. 3 The direct relationship between aging and telo- mere length
Since the telomere theory of aging suggests that cellu- lar senescence results from the curtailing of telomeres, one would expect that the prevention of telomere short- ing and novel mechanisms to to extend the length of te- lomeres would prevent or reverse the aged, or atleast
prevent the upbringing of cellular senescence. Research has suggested that this theoretical prevention of cellular senescence is indeed the case. Telomerase, a recently dis- covered enzyme that adds DNA sequences to the ends of telomeres, initiates cell division if it is inserted into cul- tured cells that have reached the Hayflick limit. Although telomerase appears to reverse cellular senescence and allows unlimited cell division, the enzyme also appears in cancerous cells: the same enzyme reversing the aging of cells has also been hypothesized to bring about the dead- ly outcontrolled cell divison known as cancer. Nonethe- less, telomerase appears to be a feasible and viable option to delay or reverse aging, as discussed next.
Insight on future therapies to combat the deleterious and unwanted aging process has been provided by numerous biogerontology groups. Telomerase, an enzyme discussed in the previous section, has been proposed as an antiag- ing breakthrough, reasoning that since it prevents aging in cells, it should prevent aging in humans as well. Future telomerase therapies involve the activation of telomerase promoter sites. Activating such promoter sites will in- crease telomerase levels within the cells, and, according to the telomere theory, delay or reverse aging. Such could possibly be perpetrated with gene therapy, although the technology is still within its infantile stage. TA-65, a telo- merase promoter, seems to be a likely possible target for gene therapy. Antiaging companies such as Sierra Sciences have found dozens of other telomerase active sites which could possibly be utilized to prevent aging. However, there is still much to research to do and drugs take years to develop. Even so, prospective telomerase human therapies to combat aging have been hypothe- sized to emerge in approximately 20 years or less.
Anotheri antiaging treatment is caloric restriction. Cur- rently, caloric restriction cannot be obtained through therapies, but rather through a diet restriction. The idea of caloric restriction is to basically reduce calorie intake while maintaining an organism’s nutritional needs. Such has been shown to increase lifespan in nematode worms, flies, and mice. Not only do the the laboratory animals live longer under a caloric restricted diet, but they also seem to age more slowly and function as though the or- ganism were still in its youth. From the free radical theory of aging, we were able to derive the fact that aging occurs due to the accumulation of free radicals and ROS mole- cules throughout cells. By restricting the number of calo- ries consumed, there is less opportunity for such toxic molecules to accumulate. Although the mechanism by which caloric restriction acts is not yet fully understood, researchers have attributed the antiaging effects of caloric restriction from decreased rates of cell division, slower metabolism, and mitigated free radical production (due to decreased phosphorylation). A possibility to utilize the life-extending characteristics of caloric restriction is to
IJSER © 2010 http://www.ijser.org
4 International Journal of Scientific & Engineering Research, Volume 2, Issue 1, January-2011
ISSN 2229-5518
identify the genes which act through the caloric restric- tion pathway, and then control the activity of the genes through gene therapy to promote an extended lifespan and delay the process of aging.
Recent research has supported the telomere and DNA damage theories of aging. A team of researchers at the Spanish National Cancer Centre, led by Maria A. Blasco, genetically engineered mice to produce 10 times the nor- mal levels of telomerase. The result was the mice living
50% longer than normal lived controls. Even more recent- ly, a group of scientists activated a telomerase control gene in mice, and were able to successfully reverse the detrimental effects of aging.
Research regarding the free radical theory of aging has been supported and yet remains inconclusive. As it is already known, caloric restriction yields a lifespan. Such an effect has been observed in nematode worms, flies, and mice. In the nematode worm C. elegans, lifespan has been shown to be extended by over 50% through caloric restriction. While this evidence partly supports the free radical theory of aging, other research suggests otherwise. Some common antioxidants are the vitamins A, C, and E. Contrary to the implications of the theory, increasing the doses of such antioxidants in laboratory animals does not increase lifespan. In fact, it actually reduces lifespan by a diminutive amount. However, one notable antioxidant, resveratrol, has been shown to extend lifespan in yeast, worms, and flies. Although the effects of antioxidants still remain controversial, they still seem to be promising tools in the area of life-extension.
The free radical, telomere, and DNA damage theories of aging have all been deliberately discussed throughout this review. Possible life-extension methods have also been noted. Although these damage-based aging theories still remain theories and not tangible facts, evidence has suggested that the accumulation of damage, the basis of all these theories, do infact, function aggregate as a result of aging. Biogerontology remains an active area of re- search today, and still, not one knows the true cause of biological senescence. Aging may result from damage- based theories or programmed-based theories, or even more likely, a combination of both. Even though numer- ous aging mechanisms still remain to be elucidated, it is anticipated by a number of experts that aging will be cured in the next 20 years. Indeed, with the current rate of scientific research, biological immortality will soon be achieved, and the horrendous disease known as aging will soon be regarded as facet of humanity’s dismal past.
The author wishes give thanks to professor Shah of UCSF for stimulating interest in the aging process and classmate Richard Zhang for providing continuous support.
[1] L.G. del Valle, “Oxidative stress in aging: Theoretical outcomes and clinical evidences in humans” Biomedicine and Pharmacotherapy, Sept. 2010.
[2] Lisa A. Brennan, “Mitochondrial function and redox control in the aging eye: Role of MsrA and other repair systems in cata- ract and macular degenerations,” published.
[3] Thomas C. Squier, “Oxidative stress and protein aggregation during biological aging” Experimental Gerontology, August
2001.
[4] Wulf Dröge, "Free Radicals in the Physiological Control of Cell
Function" Physiological Reviews, Janu. 2002.
[5] Bei Chen, Yi Zhong, Wei Peng, Yu Sun, Yu-juan Hu, Yang Yang and Wei-jia Kong, "Increased mitochondrial DNA damage and decreased base excision repair in the auditory cortex of d- galactose-induced aging rats" Molecular Biology Reports, No- vember 2010.
[6] Allison A. Johnson and Kenneth A. Johnson, "Exonuclease Proofreading by Human Mitochondrial DNA Polymerase" The Journal of Biological Chemistry, July 2001.
[7] McClintock D, Ratner, and Lokuge M, "The Mutant Form of Lamin A that Causes Hutchinson-Gilford Progeria Is a Bio- marker of Cellular Aging in Human Skin," published.
[8] BP Best, "Nuclear DNA damage as a direct cause of aging" Re- juvenation Research, 2009.
[9] Korf B. "Hutchinson-Gilford progeria syndrome, aging, and the
nuclear lamina". N. Engl. J. Med, 2008.
[10] Greider CW and Blackburn EH, "Identification of a specific telomere terminal transferase activity in Tetrahymena extracts". Cell, Dec. 1985.
[11] Bryan TM, Englezou A, Gupta J, Bacchetti S, and Reddel RR, "Telomere elongation in immortal human cells without detecta- ble telomerase activity".EMBO J., Sept. 1995.
[12] Antonia Tomás-Loba, Ignacio Flores, Pablo J. Fernández- Marcos, María L. Cayuela, Antonio Maraver, Agueda Tejera, Consuelo Borrás, Ander Matheu, Peter Klatt, Juana M. Flores, José Viña, Manuel Serrano and Maria A. Blasco, "Telomerase Reverse Transcriptase Delays Aging in Cancer-Resistant Mice" Cell, Nov. 2008.
[13] Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadiñanos J, Horner JW, Maratos-Flier E, and Depinho RA, "Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice." Nature, Nov. 2010.
[14] Saito, K., Yoshioka, H., and Cutler, R. G. "A spin trap, N-tert- butyl-alpha-phenylnitrone extends the life span of mice." Biosci Biotechnol Biochem, 1998.
[15] Howitz KT, Bitterman KJ, and Cohen HY, "Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan"
Nature, Sept. 2003.
IJSER © 2010 http://www.ijser.org