International Journal of Scientific & Engineering Research Volume 2, Issue 7, July-2011 1
ISSN 2229-5518
Biological Sequence Matching Using Fuzzy Logic
Nivit Gill, Shailendra Singh
Abstract: Sequence alignment is the most basic and essential module of computational bio-informatics. In this paper, we propose a multiple sequence alignment algorithm that employs fuzzy logic to measure the similarity of sequences based on fuzzy parameters. To guarantee the optimal alignment of the sequences, dynamic programming is used to align the sequences. The algorithm is tested on few sets of real biological sequences taken from NCBI bank and its performance is evaluated using SinicView tool.
Index Terms: Biological sequences, Dynamic programming, Fuzzy logic, Fuzzy matching score, Fuzzy parameters, Global alignment, Multiple sequence alignment.
—————————— • ——————————
enetic code of every organism can be represented as a sequence of alphabets, such as four base pairs of DNA and RNA, or twenty amino acids of protein. Over time,
these biological sequences undergo changes, called mutations, and as a result, many organisms evolve. All living organism cell are composed of DNA molecules that are passed from one generation to other. This is the reason for some living organisms being biologically similar and some being distinct. The goal of bioinformatics is to align a large number of sequences in order to study their evolutionary relationships through comparative sequence analysis.
With the help of bio-informatics, computations are applied to the biological sequences in order to analyze and manipulate them. The prime objective behind this is to discover and record the role of genetics in an organism’s biological characteristics. Sequence alignment is the most basic and essential part of computational bio-informatics and provides a base for other tasks of bioinformatics, such as sequence assembly, sequence annotation, structural and functional prediction, evolutionary or phylogeny relationship analysis. A sequence alignment refers to the method of arranging biological sequences in order to search similar regions in the sequences. The sequences with high degree of similarity have similar structure and function, and such sequences help in deriving evolutionary or phylogenetic relationships among organisms.
In this paper, we propose to align multiple biological sequences by matching the sequences using fuzzy logic. In the proposed method, the given biological sequences are compared pair wise so as to determine the number of matches, and mismatches between them. Then these counts are fuzzified using fuzzy membership functions, and then fuzzified counts are put in an aggregate fuzzy function in order to find the fuzzy match value of the two sequences. The fuzzy match value is further used to order the
————————————————
Shailendra Singh is an Assistant Professor in the department of
sequences according to the similarity. The most similar pair is aligned first and the rest of the sequences are then aligned progressively, to this aligned pair.
The outline of this paper is: Section 2 discusses the basics of sequence alignment and its types and Section 3 reviews the related work. The fuzzy logic concepts and its usage in the proposed algorithm are provided in Section 4. Section 5 details the classical Needleman-Wunsch algorithm. The proposed algorithm is described in Section 6. Experimental results and their discussions are presented in Section 7 and finally Section 8 concludes the paper.
Any biological sequence is formed from a sequence of characters drawn from an alphabet. For DNA sequence, the character alphabet is {A, C, G, T}, for RNA sequence, the alphabet is {A, C, G, U}, and for protein sequence, the character set is {A, R, N, D, C, Q, E, G, H, I, L, K, M, F, P, S, T, W, Y, V}. A sequence alignment is the process of identifying one-to-one correspondence among sub-units of sequences in order to measure the similarities among them. These similar regions provide functional, structural, and evolutionary information about the sequences under study. Aligned sequences are generally represented as rows within a matrix. Gaps (‘-‘) are inserted between the characters so that identical or similar characters are aligned in successive columns. Gaps are also called indels, as they represent insertion of a character in or a deletion of a character from a biological sequence. Sequence alignment of two biological sequences is called pair-wise sequence alignment, and in case more than two biological sequences are involved, it is called multiple sequence alignment [12]. The sequence alignment can be global or local. Global alignment "forces" the alignment to span the entire length of all query sequences (Figure 1). Local alignments identify regions of similarity within long sequences that are often widely divergent overall (Figure 2).
computer science and engineering, Punjab Engineering College-
IJSER © 2011
University of Technology, Chandigarh, India. E-mail: http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 2, Issue 7, July-2011 2
ISSN 2229-5518
![]()
Fig. 1 Global Alignment of two biological sequences
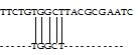
Fig. 2 Local Alignment of two biological sequences
Scoring Matrices are used to quantify the similarity achieved by an alignment [12]. These matrices contain a value (positive, zero or negative value) for each possible substitution, and the alignment score is the sum of the matrix's entries for each aligned pair. For gaps (indels), a special gap score is used--a very simple one is just to add a constant penalty score for each indel. The optimal alignment is the one which maximizes the alignment score. Commonly used matrices are PAM (Percent Accepted Mutations) matrices, BLOSUM (BLOck SUbstitution Matrix), etc. The most popular algorithms for local and global alignment are based on dynamic programming: Needleman-Wunsch algorithm for global alignment, and Smith-Waterman algorithm for local alignment.
As the sizes of biological sequence databases grow exponentially, the need for fast and efficient sequence alignment algorithms is ever-increasing. Most of the research work has been intended on primarily providing new algorithms with the main requisite of the meeting the demands of efficient sequence alignment. Researchers have used all the latest techniques with the aim of providing fast and efficient alignment algorithms.
Needleman and Wunsch proposed a dynamic programming algorithm for performing a global alignment of two sequences [1]. Smith and Waterman proposed an algorithm to find a pair of segments one from each of two long sequences such that there is no other pair of segments with greater similarity (homology) [2]. In this local alignment algorithm, similarity measure allowed arbitrary length deletions and insertions. A new algorithm for local alignment of DNA sequences had been proposed by Das and Dey [4]. Naznin, Sarker and Essam designed an iterative progressive alignment method for multiple sequence alignment by using new techniques for both generating guide trees for randomly selected sequences as well as for rearranging the sequences in the guide trees [10]. Paul and Konar proposed direct comparison methods to obtain global and local alignment between the two sequences [5]. They also proposed an alternate scoring scheme based on fuzzy concept. Cai, Juedes, and Liakhovitch proposed to combine existing efficient algorithms for near optimal global and local multiple sequence alignment with evolutionary computation techniques to search for better near optimal sequence
alignments [3]. Y. Chen et.al proposed a partitioning approach, based on ant-colony optimization algorithm that significantly improved the solution time and quality by utilizing the locality structure of the problem [6]. Yue and Tang applied the divide-and-conquer strategy to align three sequences so as to reduce the memory usage from O (n3) to O(n2). They used dynamic programming so as to guarantee optimal alignment [9]. Nasser et al. provided a hybrid approach of dynamic programming and fuzzy logic to align multiple sequences progressively [8]. They computed optimal alignment of subsequences based on several factors such as quality of bases, length of overlap, gap penalty. Chang et al. established fuzzy PAM matrix using fuzzy logic and then estimated score for fitness function of genetic algorithm using fuzzy arithmetic [7]. Their experimental results evidenced fuzzy logic useful in dealing with the uncertainties problem, and applied to protein sequence alignment successfully.
In all this work, the main objective of the researchers had been to apply different techniques in order to provide efficient alignment algorithms in terms of time and memory requirements.
Fuzzy logic is based on the fuzzy-set theory proposed by L.A. Zadeh in 1965. It is a form of many-valued logic which deals with reasoning that is approximate rather than fixed and exact. In contrast with "crisp logic", where binary sets have two-valued logic: true or false, fuzzy logic variables may have a truth value that ranges in degree between 0 and
1 [16]. Fuzzy logic has been extended to handle the concept of partial truth, where the truth value may range between completely true and completely false.
In a fuzzy system, the values of a fuzzified input execute all the rules in the knowledge repository that have the fuzzified input as part of their premise. This process generates a new fuzzy set representing each output or solution variable. Defuzzification creates a value for the output variable from that new fuzzy set [11]. So, in order to apply fuzzy logic to an application, first the inputs must be fuzzified so that their value is in the range 0 to 1, then the rules defined by the application are applied, and after this, the results derived from various rules are combined using an aggregation function. Finally, the aggregated results are defuzzified by using an inference function. The evaluations of the fuzzy rules and the combination of the results of the individual rules are performed using fuzzy set operations. The operations on fuzzy sets are different than the operations on non-fuzzy sets [13]. The operations for OR and AND operators are max and min, respectively. For complement (NOT) operation, NOT(A) is evaluated as (1-A).
The proposed sequence-matching algorithm uses three input variables – match-count (#match), mismatch-count (#mismatch), and calculated-score (#score – calculated using substitution matrix). These inputs are then fuzzified using following membership functions:
IJSER © 2011 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 2, Issue 7, July-2011 3
ISSN 2229-5518
0, if #match = 0,
Jl(match) = 1, if #match = lenSeq, (1)
[0,1] (#match / lenSeq)
0, if #mismatch = 0,
Jl(mismatch) = 1, if #mismatch = lenSeq, (2)
[0,1] (#mismatch / lenSeq)
0, if #score :5 0
Jl(score) = 1, if #score = perfectScore, (3)
[0,1] #score/perfectscore
In these equations, lenSeq is the length of the shorter
sequence of the two sequences being matched, and
perfectScore is the score of matching the two candidate
sequences, if there are no indels or replacements.
In Needleman-Wunsch algorithm, a scoring matrix is calculated for the two given sequences A and B, by placing one sequence along row side and another column side. The size of the matrix is (M+1)*(N+1) (M and N are the lengths of the two sequences). The optimal score at each matrix (i, j) position is calculated by adding the current match score to previously scored positions and subtracting gap penalties, which may evaluate to either a positive, negative or 0 value. A matrix F(i, j) indexed by residues of each sequence is built recursively, such that
F(i, 0) = F(0, j) = 0
F(i, j) = max { F(i-1, j-1) +S(xi, yj), F(i-1, j) + G, F(i, j-1) + G }
subject to boundary conditions; here, S(i, j) is the
substitution score for residues i and j, and G is the gap
penalty [17].
The proposed algorithm uses the Needleman-Wunsch
algorithm for aligning two biological sequences. The
substitution matrix S, given in Figure 3, is used to calculate
the scores.
A C G T A 2 -1 1 -1
C -1 2 -1 1
G 1 -1 2 -1
T -1 1 -1 2
Fig. 3 Substitution Matrix used in the proposed algorithm
An alignment is computed using the F-matrix (calculated above): start from the bottom right cell, and compare the cell value with the three possible sources ((i-1, j-1) i.e. a Match, (i, j-1) i.e. an Insert, and (i-1, j) i.e. a Delete) to see which it came from. If it is same as Match, then Ai and Bj are aligned, if same as Delete, then Ai is aligned with a gap, and if same as Insert, then Bj is aligned with a gap.
The proposed algorithm attempts to align multiple biological sequences (DNA, for example), using fuzzy logic. The algorithm uses progressive approach for aligning
multiple sequences, by first aligning the two most similar sequences, using Needleman-Wunsch algorithm. Then sequences are chosen, one by one, from the remaining sequences, and are aligned to the aligned set of sequences. The proposed algorithm is composed of following two algorithms:
This algorithm finds the fuzzy score of similarity, based on fuzzy parameters (as given in section 4), for the given two biological sequences A and B.
I. Calculate the number of matches and mismatches between the two sequences A and B. Also calculate the score using the substitution matrix.
II. Fuzzify the MatchCount, MismatchCount, and Score into the µ(match), µ(mismatch), and µ(score) using the equations 1, 2 and 3, given in section 4.
III. Calculate the aggregate fuzzy_similarity_score based on the three fuzzy parameters.
This algorithm aligns the given set of N sequences stored in SeqDB using progressive approach. The resultant aligned sequences are stored in AlignDB.
I. Find the fuzzy similarity value between all pairs of sequences using algorithm MATCH_SEQFL. Remove the most similar pair of sequences from the input set, say it is sequences P and Q. Align sequences P and Q using the Needleman Wunsch algorithm. Say, the corresponding aligned sequences are AlignedP and AlignedQ. Store these aligned sequences in AlignDB.
II. Select a sequence R from SeqDB, which has higher fuzzy similarity value to one of the aligned sequences. Now align the sequence R with the aligned sequences AlignedP and AlignedQ. Find the fuzzy match value of both aligned pairs, using MATCH_SEQFL algorithm. From the two aligned versions of R, select the one which gives higher fuzzy similarity score. Say, it is alignR, and store it in AlignDB. Remove the sequence R from the given sequence set SeqDB.
III. Repeat Step II, until all the biological sequences of
SeqDB have been aligned.
MATLABTM was used to implement the proposed algorithm. The fuzzy logic toolbox GUI tool provides an easy method to build a fuzzy inference system [15]. Using this tool, we designed a fuzzy sequence matcher system for implementing the algorithm (Fig. 4).
IJSER © 2011 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 2, Issue 7, July-2011 4
ISSN 2229-5518
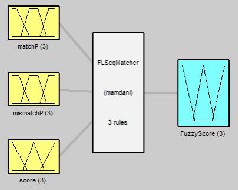
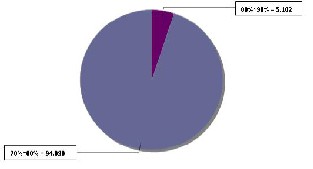
Fig. 5(a) Detailed text view of the AH1N1 sequence-set alignment results for the (401-500) base pairs
Fig. 4 System FLSeqMatcher with 3 inputs, 1 output, and 3 rules
Three different sets of influenza virus genome sequences (host: human): AH1N1, AH1N2, and AH1N3, for different countries were collected (randomly) from NCBI’s Influenza virus resource site [18]. Table I enlists the various attributes of the tested sequence sets.
The proposed algorithm was used to align all the three sets of sequences. The performance of the alignment results was evaluated using SinicView – a visualization environment for comparison of multiple sequence alignment tools [14].
TABLE I
TESTED SEQUENCE SETS
Influenza Virus type | No. of sequen-- ces | Average Length (bp) | Origin (Country/ Continent) | Collection Year |
AH1N1 | 34 | 1075 | USA | 2007-2009 (majorly 2007) |
AH1N2 | 17 | 872 | Asia | 2007-2009 |
AH3N2 | 50 | 1033 | USA | 2008 |
The alignment results for AH1N1, AH1N2 and AH3N2 virus sets are shown in figures 5(a), 6(a) and 7(a) respectively. The line graph (in red) shows the percent identity plot for the whole length of sequences. The second portion in these figures shows the detailed text alignments for the base pairs ranging from 401 to 500. The left side of the second portion lists the names of the sequences, and the right side shows the corresponding aligned sequences. The vertical red bar indicates an identity match in all the sequences. Fig. 5(b), 6(b), and 7(b) plot the distribution of percent identical rate of the three aligned sequence- sets respectively.
Fig. 5(b) Distribution of Per cent Identical rate in the aligned AH1N1 sequence-set
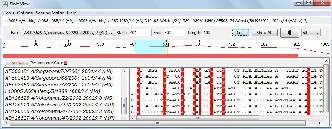
Fig. 6(a) Detailed text view of the AH1N2 sequence-set alignment results for the (401-500) base pairs
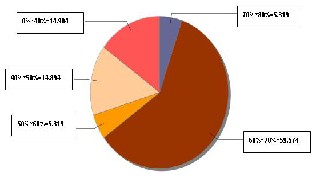
Fig. 6(b) Distribution of Per cent Identical rate in the aligned AH1N2 sequence-set
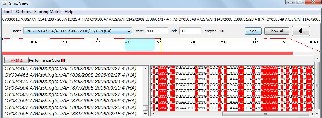
Fig. 7(a) Detailed text view of the AH3N2 sequence-set alignment results for the (401-500) base pairs
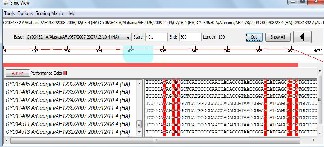
IJSER © 2011 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 2, Issue 7, July-2011 5
ISSN 2229-5518
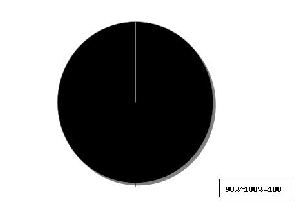
obtained clearly indicated that the algorithm based on fuzzy logic is an efficient method for biological sequence matching.
Fig. 7(b) Distribution of Per cent Identical rate in the aligned AH3N2 sequence-set
Table II summarizes the distribution of percent identical rate of alignments of candidate influenza virus sequence-sets.
TABLE II
TABULATION OF PER CENT IDENTICAL RATE DISTRIBUTION IN THE ALIGNED CANDIDATE SEQUENCE-SETS
Sequence Set | Per cent Identical Rate | Distribution % |
AH1N1 | 70 ~ 80 | 94.898 |
AH1N1 | 80 ~ 90 | 5.102 |
AH1N2 | 0 ~ 40 | 14.904 |
AH1N2 | 40 ~ 50 | 14.894 |
AH1N2 | 50 ~ 60 | 5.319 |
AH1N2 | 60~70 | 59.574 |
AH1N2 | 70~80 | 5.319 |
AH3N2 | 90 ~ 100 | 100 |
The evaluation results depict a lot of variation which can be due to the difference in the origin of sequence (i.e. country/continent) and the time (year) of collection. In the first set (AH1N1), the percent identity rate varies majorly in
80% - 90% range, as the sequences originate from USA and collection years are 2007-2009 (majority 2007). Second set’s per cent identity rate is low as the aligned sequences belong to different countries in a continent and the collection year is also different. AH3N2 sequences show high almost 100% identity rate as the sequences have been majorly collected from Washington (USA) and in the year 2008.
In this paper, an algorithm for sequence matching based on fuzzy logic has been proposed and implemented. The fuzzy similarity score of two sequences was calculated based on few fuzzy parameters. The calculated similarity score guides in the progressive alignment of multiple sequences, which was done using dynamic programming. The experimental results show that the algorithm performs the alignment of sequences quite well and the nature and relationship of sequences reveal the alignment performance. The results
[1] S. B. Needleman and C. D. Wunsch, "A General Method Applicable to the Search for Similarities in the Amino Acid Sequence of two Proteins," J. Molecular Biology, vol. 48, pp. 443-453, 1970.
[2] T. F. Smith and M. S. Waterman, "Identification of Common Molecular
Subsequence," J. Molecular Biology, vol. 147, pp. 195-197, 1981.
[3] L. Cai, D. Juedes, E. Liakhovitch, “Evolutionary Computation Techniques for Multiple Sequence Alignment”, Proceedings of the 2000 Congress on Evolutionary Computation, 2000, pp. 829-835
[4] Swagatam Das & Debangshu Dey, “A New Algorithm for Local
Alignment in DNA Sequencing”, Proc. of IEEE Conference, INDICON
2004, pp. 410-413
[5] Bandyopadhyay, S.S.; Paul, S.; Konar, A., “Improved Algorithms for DNA Sequence Alignment and Revision of Scoring Matrix”, Proceedings of International Conference on Intelligent Sensing and Information Processing, 2005, pp. 485-490
[6] Y. Pan, Y. Chen, Juan Chen, Wei Liu, Ling Chen “Partitioned Optimization Algorithms for Multiple Sequence Alignment”, Proceedings of the 20th International Conference on Advanced Information Networking and Applications, 2006, pp. 5
[7] Pin-Teng Chang, Lung-Ting Hung, Kuo-Ping Lin, Chih-sheng Lin, Kuo- Chen Hung, “Protein Sequence Alignment Based on Fuzzy Arithmetic and Genetic Algorithm”, 2006 IEEE International Conference on Fuzzy Systems, pp. 1362-1367
[8] Sara Nasser, Gregory L. Vert, Monica Nicolescu1 and Alison Murray, “Multiple Sequence Alignment using Fuzzy Logic”, Proceedings of the
2007 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology, pp. 304-311
[9] Feng Yue and Jijun Tang, “A Divide-and-Conquer Implementation of Three Sequence Alignment and Ancestor Inference”, 2007 IEEE International Conference on Bioinformatics and Biomedicine, pp. 143-150
[10] Farhana Naznin, Ruhul Sarker, and Daryl Essam, “Iterative Progressive Alignment Method (IPAM) for Multiple Sequence Alignment”, Computers & Industrial Engineering, 2009. pp. 536-541
[11] E. Cox, “Fuzzy Fundamentals”, IEEE Spectrum October 1992, Volume 29, Issue 10, pp 58-61
[12] David W. Mount, “Bioinformatics: Sequence and Genome Analysis”, Cold
Spring Harbor Laboratory Press
[13] “A Short tutorial on Fuzzy Logic”, http://www.cs.bilkent.edu.tr/~bulbul/depth/fuzzy.pdf
[14] http://biocomp.iis.sinica.edu.tw/new/SinicView_tr05005.pdf
[15] http://www.mathworks.com/help/toolbox/fuzzy/ [16] http://en.wikipedia.org/wiki/Fuzzy_logic
[17] http://en.wikipedia.org/wiki/Needleman-Wunsch_algorithm
[18] http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi
IJSER © 2011 http://www.ijser.org