International Journal of Scientific & Engineering Research, Volume 4, Issue 4, April-2013
ISSN 2229-5518
THEORETICAL INVESTIGATION OF THE ELECTRONIC PROPERTIES OF ZIGZAG
1695
GRAPHENE NANORIBBONS WITH PYRIDINE AND PORPHYRIN-LIKE DEFECTS
Michael Mananghaya
—————————— • ——————————
ingle layer of graphite called graphene has attracted con- siderable research interests owing to its novel properties. Physical properties such as massless Dirac fermions behav-
ior [1-5], room-temperature quantum hall effect [6, 7] and high mobility and coherence [8] open up new perspectives and fu- ture research. The two-dimensional (2D) graphene sheet itself is a semi-metal. However, when the 2D sheet is cut into rec- tangle slices, namely, graphene nanoribbons (GNRs), they can become one-dimensional (1-D) semiconductors with their en- ergy band gap depending on the width and crystallographic orientation of cutting edge of the nanoribbons. The GNRs could exhibit different edge geometries including zigzag and armchair, which will determine their electronic structures. In particular, the confinement of the electronic wave functions and the presence of the edges will open a band gap, which will make them suitable for the applications in semiconductor de- vices. Therefore, GNRs appear to be excellent candidates for the design of novel semiconductor devices [9-20].
Several recent theoretical studies have also revealed that zigzag edged GNRs (ZGNRs) can be converted into a half metal by either applying an external electric field or through chemical modification of the edges. Half metals hold the promise for spintronic applications [18, 21-27] as the electric current can be fully spin polarized when going through half metals. This is because for a half metal, one electron spin channel is insulating while the other channel is metallic. The spin density of states of functionalized GNRs doped with ni- trogen (N) atoms was calculated using unrestricted density functional theory (DFT) and it was found out that that the
edge substitutions at low density do not remarkably change the band gap, whereas the bulk substitution of N atoms will promote the onset of semiconductor–metal transitions. Aside from edge substitutions N dopants can also be substituted into the C network with a vacancy formation. Porphyrin-like (labeled as 4ND) and pyridine-like defects (labeled as 3NV) are formed in ZGNR as verified through spectroscopic measurements. ZGNR decorated with NO2 chemical function group could become half metallic [26]. Chemical modification can elevate certain spin-polarized bands crossing the Fermi level. However, the large steric interactions between neighbor groups make the system distorted. Hence, our design is mainly focused on sin- gle-atom for the chemical functionalization.
The electronic properties were studied using first-principles density function theory (DFT) calculations are carried out via Dmol3 package [27-29]. The generalized gradient approxima- tion (GGA) in the Perdew-Burke-Ernzerhof (PBE) form and an all-electron double numerical basis set with polarized function (DNP basis set) were chosen for the spin-unrestricted DFT computation [30] set with atomic cutoff at 5.5 Å. To simulate ZGNRs, a cuboid supercell was used with a wall-to-wall dis- tance of at least 20 Å sufficient enough to avoid in-plane inter- actions between adjacent cells. For geometric optimization, the Brillouin zone was sampled by 3 k points using the Monk- horst-pack scheme [31], the forces on all atoms were optimized to be less than 0.005 eV/Å using the Broyden-Fletcher- Goldfarb-Shanno (BFGS) algorithm.
————————————————
• Michael Mananghaya is a research scholar at De La Salle University, Phil- ippines. mikemananghaya@gmail.com
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 4, Issue 4, April-2013
ISSN 2229-5518
1696
(a)
(b)
(c)
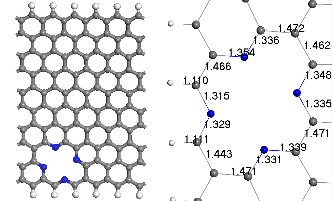
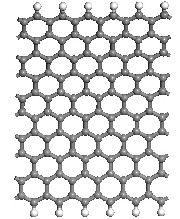
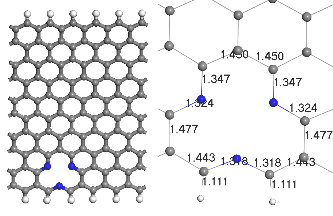
Fig.1. Optimized geometry of (a) the infinite ZGNR, (b) ZGNR with pyridine defects (c) ZGNR with porphyrine defects. Gray color depicts carbon atoms blue is Nitrogen and white is Hydrogen.
Two types of modified ZGNRs were considered: Pyridine and porphyrin defects usually occur in graphene-based sys- tems. 3NV defect can be constructed by removing a C atom and substituting the three nearest neighbor C atoms with N atoms (3NV-ZGNR) as seen in Fig. 1(b), substitution of nitro- gen dopants with vacancy formation, by removing two C atom among four hexagons and replacing the four surround- ing C atoms with 4 N atoms (4ND-ZGNR) as seen in Fig. 1(c)
3.1. Structure of 10-ZGNR, 3NV-ZGNR and 4ND-ZGNR
Fig. 1(a) shows the relaxed geometries of the nanoribbon, which was chosen as a typical semiconducting model for in- vestigation. The width of the pure ZGNR was defined with the width parameter Nz of zigzag grapheme nanoribbons as the number of zigzag lines across the ribbon width, as exem- plifyied by 10-ZGNR was chosen in the present work. The edge carbon atoms of the nanoribbon are all saturated with H atoms to avoid the dangling bond states. The total energies for different magnetic phases of 10-ZGNR, including nonmag- netic (NM), ferromagnetic (FM), and antiferromagnetic (AFM) were calculated to determine the ground state. The total en- ergy of the FM phase is lower than that of the NM phase but higher than that of the AFM phase. Therefore the AFM phase is the most favorable for 10-ZGNR. The results are in agree- ment with literature, indicating that the methods in this work are reliable to describe fully the electronic properties of ZGNRs.
Due to the two missing C atom(s), the C–N bond lengths of pyridine-like and porphyrin-like doping are deter- mined to be ~1.318–1.347 Å (see Fig. 1(b)) and ~1.315–1.354 Å (see Fig. 1(c)) respectively, depending on the orientation, as compared to ~1.407-1.443 Å for the C–C bonds. Thus, N impu- rities in ZGNR s produce their own local strains and deforma- tion.
3.2. Electronic properties of 10-ZGNR, 3NV-ZGNR and 4ND- ZGNR
The formation energies (Ef) defined as:
Ef = Etot − nC(µC− nHµH)/120 − nNµN− nHµH, (1)
where Etot is the total energy of the 10-ZGNR with 3NV or
4ND defects, nC nN and nH are the number of C, N and H at- oms, respectively. µC is the chemical potential of C obtained from the corresponding pure 10-ZGNR, and µN with µH is the chemical potential of N, H obtained from nitrogen and hydro- gen in gas phase. There are many possible configurations since the defect can exist in different locations but computation shows that the configuration with defects close to the edge is energetically favored. The formation energy of the 3NV defect is lower than that of the 4ND defect indicating that 3NV defect is more likely to form in 10-ZGNR. The calculated formation energy of the 3NV-ZGNR is on the average of 2.78 eV and 3.70 eV for the 4ND-ZGNR.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 4, Issue 4, April-2013
ISSN 2229-5518
1697
(a) (b) (c)
(a)
0.10
0.10
0.10 90
0.08
0.08
0.08 80
0.06
0.06
0.06 70
0.04
0.04
0.04
60
0.02
0.02
0.02
50
0.00
-0.02
-0.04
-0.06
-0.08
0.00
-0.02
-0.04
-0.06
-0.08
0.00
40
-0.02
30
-0.04
20
-0.06
10
-0.08
-0.10
a b c d e Z
-0.10
G a b c d e Z
-0.10
G a b c d
0
-0.9 -0.8 -0.7 -0.6 -0.5 -0.4 -0.3 -0.2 -0.1 0.0 0.1 0.2
(b)
s p
alpha beta 24
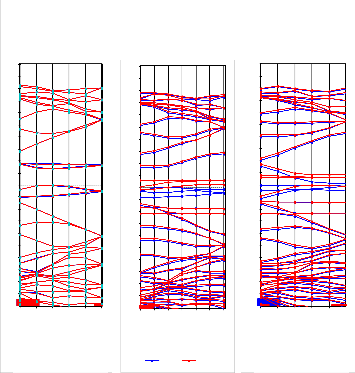
Fig.2. The band structures of (a) pure 10-ZGNR, (b) 10-ZGNR 22
with pyridine-like defect and (c) 10-ZGNR with porphyrin-
20
like defect, Fermi level is set as zero and plotted with a bro-
ken line. 18
16
To clarify the issues on the effects of the formation of 3NV 14
and 4ND defects have on the electronic properties of ZGNR
12
the band structures of the ZGNR with 3NV and 4ND defects
are examined in Fig. 2. In Fig. 2(a) the spin resolved band 10 structures of 10-ZGNR is shown. The spin-up and spin-down 8 bands are fully degenerate, the net magnetic moment of the 6
AFM phase is zero and a 0.35 eV energy gap is observed. After
4
the introduction of a 3NV defect, the energy bands near the
Fermi level become asymmetric for different spin channels: in 2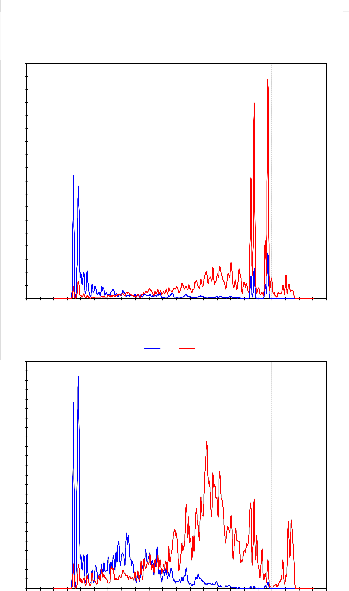
the spin-up channel, the Fermi level is shifted into the conduc- 0
tion band and metallicity is thus obtained, while the spin- down channel is semiconducting. Therefore, the 10-ZGNR containing a 3NV defect is half-metallic. Similarly, the 10- ZGNR containing a 4ND defect is also half metallic, with a metallic spin-down channel and semiconducting for the spin- up channel. The results demonstrate that the introduction of pyridine and porphyrin defects in GNRs can lead to the break- ing of spin degeneracy, suggesting a feasible way of building spin devices based on GNRs.
The formation of pyridine-like nitrogen defects is very cru- cial for enhancing the metal binding to the defects. The strong interactions between the N and the ZGNR with defects can be explained through partial densities of states (PDOS), that is, N adsorption on the 3NV-ZGNR as shown in Figs. 3(a) and 3(b) for the 10-ZGNR. It is found that the p electrons of C and N atoms mainly contribute to the electronic states near Fermi level. In other words, strong interaction exists between the p orbitals of N and atoms due to their hybridization with each
-0.9 -0.8 -0.7 -0.6 -0.5 -0.4 -0.3 -0.2 -0.1 0.0 0.1 0.2
Energy (Ha)
![]()
![]()
s p
Fig.3. The PDOS of (a) N and (b) C in the adsorption system for the
10-ZGNR. The blue and red plots denote s and p orbitals, respectively.
each other. The ZGNR with 3NV defects uses two valence electrons to form a lone pair. The reactivity of the 3NV-ZGNR is greatly enhanced compared with the case of the pure ZGNR due to the non-bonding lone pair.
The electronic properties of zigzag graphene nanoribbons with pyridine and porpyrine defects were studied using spin- unrestricted density functional theory. The formation of con- figuration with pyridine and porphyrin defects close to the edge is energetically favored and thermodynamically stable. The calculated band structures and density of states denote
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 4, Issue 4, April-2013
ISSN 2229-5518
1698
that the impurities can impose a significant effect on the elec- tronic properties of 10-ZGNR. Pyridine and porphyrin defects in ZGNR caused a semiconductor to half metallic transitions. Finally, the computations point out a feasible way for achiev- ing spintronic devices based on ZGNRs in the very near fu- ture. .
This work was supported by the Department of Science and Tech- nology, Philippine Council for Industry, Energy and Emerging Technology Research and Development (PCIEERD) formerly Phil- ippine Council for Advanced Science and Technology Research and Development (DOST-PCASTRD) for the acquisition of the Dmol3 v6.0 software. Also, this was supported in part by the Science Education Institute (DOST-SEI) and the Physics and Chemical En- gineering Department of De La Salle University-Manila.
[1] K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, I.V. Katsnelson, I. V.
Grigorieva, S. V. Dubonos, A. A. Firsov, "Two-dimensional gas of massless
Dirac fermions in grapheme,” Nature, vol. 438, pp. 197, 2005.
[2] M. L Katsnelson, K. S. Novoselov, A. K. Geim, “Chiral tunnelling and the
Klein Paradox in Graphene,” Nature Physics, 2006, vol. 2, pp. 620, 2006.
[3] A. H. Castro Neto, F. Guinea, N. M. R. Peres, “Drawing conclusions from graphene,” Physics World, vol. 19, pp. 33, 2006.
[4] S. Y. Zhou, G. H. Gwenon, J. Graf, A. V. Ferdorov, C.D. Spataru, R. D. Diehl, Y. Kopelevich, D. H. Lee, S. G. Louie, A. Lanzara, Nature Physics, vol. 2, pp.
595-599, 2006.
[5] A. K. Geim, K. S. Novoselov, “The Rise of Graphene,” Nature Materials, vol. 6 (3), pp. 183-191, 2007.
[6] K. S. Novoselov, Z. Jiang, Y. Zhang, S. V. Morozov, H. L. Stormer, U. Zeit- ler, J.C. Maan, G. S. Boebinger, P. Kim, A. K. Geim, “Transparent and con- ducting electrodes for organic electronics from graphene oxide,” Science, vol.
315, pp. 1379, 2007.
[7] Y. Zheng, T Ando, “Hall conductivity of a two-dimensional graphite system,” Physics Review B, vol. 65, ID 245420, 2002.
[8] K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Du- bonos, A. A. Firsov, “Electric Field Effect in Atomically Thin Carbon Films,” Science, vol. 306 (5696), pp. 666-669, 2004.
[9] M. Fujita, K. Wakabayashi, K. Nakada, K. J. J. Kusakabe, “Peculiar Localized
State at Zigzag Graphite Edge,’” Physics Society Japan, vol. 65, pp. 1920–
1923, 1996.
[10] Y. Miyamoto, K. Nakada, M. Fujita, “Electronic and magnetic properties of oxygen patterned graphene superlattice,” Pysics Review B, vol. 60, 16211,
1999.
[11] K. Nakada, M. Fujita, G. Dresselhaus, M. S. Dresselhaus, “Spatially resolv- ing edge states of chiral graphene nanoribbons,” Physics Review B vol. 54, pp. 17954–17961, 1996.
[12] K. Kusakabe, Maruyama, “Theoretical Prediction of Synthesis Methods to
Create Magnetic Nanographite,” Material Physics Review B, vol. 67, 092406,
2003.
[13] H. Lee, Y. W. Son, N. Park, S. Han, J. Yu, “Extinction of ferromagnetism in highly ordered pyrolytic graphite by Annealing,” Physics Review B, vol. 72,
174431, 2005.
[14] E. Rudberg, P. Salek, Y. Luo, “Half-Metallicity in Edge-Modified Zigzag
Graphene Nanoribbons,” Nano Letters, vol. 7, pp. 2211–2213, 2007.
[15] L. Pisani, J. A. Chan, B. Montanari, Harrison, “Electronic and Magnetic Prop- erties of Graphitic Ribbons,” Nano Material Physics Review B, vol. 75,
064418, 2007.
[16] O. Hod, V. Barone, G. E. Scuseria, “Half-metallic graphene nanodots: A comprehensive first-principles Theoretical Study,” Physics Review B, vol. 77, No. 035411, 2008.
[17] O. Hod, J. E. Peralta, G. E. Scuseria, “Electromechanical Properties of Sus- pended Graphene Nanoribbons,” Physics Review B, vol. 76, No. 233401,
2007.
[18] O. Hod, V. Barone, J. E. Peralta, G. E. Scuseria, “Enhanced Half-Metallicity in
Edge-Oxidized Zigzag Graphene Nanoribbons,” Nano Letters, vol. 7, pp.
2295–2299, 2007.
[19] V. Barone, O. Hod, G. E. Scuseria, “Electronic Structure and Stability of Semi- conducting Graphene Nanoribbons,” Nano Letters, vol. 6, pp. 2748–2754,
2006.
[20] Y. W. Son, M. L. Cohen, S. G. Louie, “Energy gaps in graphene nanorib- bons,” Physics Review Letters, vol. 97, No. 216803, 2006.
[21] R. A. Groot, F. M. Mueller, P. G. Engen, K. H. J. Buschow, “The surface com- position and spin polarization of NiMnSb epitaxial Thin Films,” Physics Re- view Letters vol. 50, pp. 2024, 1983.
[22] G. A. Prinz, “Magnetoelectronics,” Science, vol. 282, pp. 1660, 1998.
[23] M. Ziese, “Extrinsic magnetotransport phenomena in Ferromagnetic Ox- ides,” Reports on Progress in Physics, vol. 65, pp. 143, 2002.
[24] Y.-W. Son, M. L. Cohen, S. G. Louie, “Half-Metallic Graphene Nanoribbons,” Nature, vol. 444, 347–349, 2006.
[25] E. J. Kan, Z. Li, J. L. Yang, J. G. Hou, “Strain effect on electronic structures of graphene nanoribbons: A first Principle Study,” Applied Physics Letters, vol.
91, 243116, 2007.
[26] E. J. Kan, Z. Li, J. L. Yang, J. G. Hou, “Electronic and Magnetic Properties of Partially-Open Carbon Nanotubes,” Journal American Chemical Society, vol. 130, 4224, 2008.
[27] B. Delley, “An All-Electron Numerical Method for Solving the Local Density Functional for Polyatomic Molecules,” Journal of Chemical Physics, vol. 92, pp. 508-517, 1990.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research Volume 4, Issue 4, April-2013
ISSN 2229-5518
1699
[28] B. Delley, “From molecules to solids with the Dmol3 approach,” Journal of
Chemical Physics, vol. 113, 7756-7764, 2003.
[29] Dmol3 is a density functional theory quantum mechanical package available from Accelrys Software Inc.
[30] J. P. Perdew, K. Burke, Ernzerhof, “Generalized Gradient Approximation
Made Simple,” Materials Physics Review Letters vol. 77, pp. 3865–3868, 1996.
[31] H. J. Monkhorst, J. D. Pack, “Special points for Brillouin-zone integrations,” Physics Review B, vol. 13, pp. 5188-5192, 1976.
IJSER © 2013 http://www.ijser.org