International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 44
ISSN 2229-5518
Synthesis and spectroscopic characterization of some sterically hindered
µ-oxo bis [tricyclohexyl antimony (V)]
carboxylates and -halocarboxylates
Kiran Singhal*, Dharmendra K. Srivastava, Prem Raj
Abstract— A series of sterically hindered µ-oxo bis [tricyclohexylantimony (V)] carboxylates [(cyclo-C6H11)3Sb-O-Sb(cyclo- C6H11 ) 3 ](OCOR) 2 where -OCOR = p-trifluoromethyl mandelate, p-methoxy mandelate, salicyclate, 2pyrazine carboxylate, mandelate has been synthesised and characterised by solid state infrared, 1H , 19Fand 13C NMR spectroscopy, molecular weight and conductance measurements. The newly synthesized derivatives are monomeric in benzene and non ionic in acetonitrile with pentacoordination
dispensation around antimony.
Keywords—µ-oxo bis [tricyclohexylantimony (V)] carboxylates, halocarboxylates, IR, 1H, 19F NMR, 13CNMR spectra, monomeric, non ionic.
—————————— ——————————
Despite a considerable amount of work done on the synthesis and reactivity of organoantimony (V) carboxylates, Rn- Sb(OCOR)5-n (n= 1-4), R = alkyl, aryl, those having Sb-O-Sb framework have been studied to a limited extent [1-8]. The latter class of compounds is important in the light of fact that sodium stibogluconate (pentostam) a well known drug for the treatment of leishmainiasis have the similar framework cou- pled with gluconate constitution. Our recent in vitro an- tileishmaniasis and antimicrobial studies indicate that µ- oxobis [(triarylantimony (V)] dicarboxylates exhibit high anti-
The interaction of µ-oxy bis (tricyclohexyl antimony chloride) [(cyclo-C6 H11 )3 SbCl]2 O, with the silver salt of corresponding carboxylic acid in 1:1 and 1:2 molar ratio afforded mono and di-substituted µ-oxy bis (tricyclohexyl antimony) derivatives, respectively (Eq. 1-2).
1:1
fungal, antibacterial, antitumoral and antileishmainial activity
THF (cyclo-C H ) Sb-O-Sb(cyclo-C H )
+ AgCl (1)
[9-15]. Since the biological activity is largely affected by the nature of anion and organic group bound to the central metal atom apart from its nature of metal, geometrical dispensation around metal and oxidation state, we considered it worth-
[(cyclo-C6H11)3SbCl]2O + AgL
1:2
![]()
THF
![]()
6 11 3
![]()
Cl L
6 11 3
while to synthesize a series of µ-oxo bridged organoantimony carboxylates and –halocarboxylates having biologically poten- tial organic moieties. It may be noted that cyclohexyltin deriv- atives exhibit potential antimicrobial and antitumoral activity.![]()
[(cyclo-C6H11)3SbCl]2O + 2AgL
![]()
![]()
O
![]()
![]()
(cyclo-C6H11)3Sb-O-Sb(cyclo-C6H11)3 + 2AgCl (2)
L L
![]()
![]()
In addition to this the introduction of cyclohexyl group is L = O
F3C
![]()
![]()
OH O
![]()
![]()
C C
H3CO
![]()
![]()
![]()
![]()
OH O C C
![]()
![]()
known to render the hydrolytic stability to the system [16-23]. O H H O H O
The present investigation concerned with the synthesis of µ-
oxobis [(tricyclohexylantimony (V)] dicarboxylates and their
characterization by IR, NMR spectroscopy. Solution phase
-OCOC6H4(o-OH) 1,
![]()
![]()
OH O
-OCOCH(OH)C6H4 (p-CF3) 2
![]()
![]()
![]()
![]()
![]()
O N C
-OCOCH(OH)C6H4(p- OCH3) 3
studies complementary to above have been carried out to as-![]()
![]()
C C O
![]()
Ph O
N
-OCOC(OH)(C6H5)2 4
-OCOC4N2H3 5
————————————————
• *Corrseponding author is Dr. (Mrs.) Kiran Singhal, Associate Professor, Department of Chemistry, University of Lucknow, Lucknow, U.P., India-
226007 and is Ph. D. supervisor of Mr. D. K. Srivastava.
• Email: singhal.kiran@gmail.com, Ph: +91-9415159894
• Contract Grant Sponsor: University Grants Commission, New Delhi,
India Vide Letter No. 37-429/2009 SR.
Dr. Prem Raj is Senior Professor Chemistry Departrment, Lucknow
University, Lucknow, U. P., India.
certain the chemical behavior in solution.
As is well known α-hydroxy carboxylic acid may unclog o cyclisation using hydroxyl and carboxylic hydrogen's [23], β- hydroxy, carboxylic acids normally prefer monodentate and linear salts [24]. The only carboxylic acid undergoing cyclisa- tion was mandelic acid to form cyclic µ-oxy-bis (tricyclohexyl antimony) derivatives (7, 8), irrespective of molar ratio i.e. the same product was formed in 1:1 and 1:2 molar ratio reactions. The formation of ring compound was established on the basis of amount of residue after filtration, melting points, elemental
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 45
ISSN 2229-5518
analysis, super imposable IR spectra and other spectral analy- sis. On the other hand, the other α-hydroxy acid viz. benzilic acid, p. (trifluoromethyl) mandelic acid, p-methoxy mandelic acid do not form the cyclometallates and mono or di- substituted oxo-bridged derivatives of antimony are formed for 1:2 and 1:2 molar ratio reactions, respectively.
THF
MHz FT NMR) in solvent (DMSO + CDCl3 ) with chemical shift being reported as δ(ppm) taking tetramethylsilane as ref- erence. The peak for protons of DMSO appeared at v 83.3 ppm. The 1HNMR data of the title compounds are listed in Table-4. The chemical shifts of the protons of cyclohexyl ring appear in the range δ(1.15 – 3.80) ppm in the form of multiplet. The appearance of signal of protons of cyclohexyl ring as mul-![]()
[(cyclo-C6H11)3SbCl]2O + 2AgL/AgL
![]()
![]()
![]()
![]()
(cyclo-C6H11)3Sb O Sb(cyclo-C6H11)3 + AgCl (3)
![]()
O O O R
![]()
O
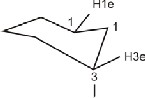
tiplet is due to low to high long range coupling .When four
sigma bonds between interacting protons adopt a 'W-
arrangement' as in the case of 1, 3 diequitorial protons of a
rigid cyclohexyl system such type of coupling are observed
(fig. 1).![]()
![]()
![]()
![]()
C O
where L = C6H5CH(OH)COO-; R = C6H5-
HO C H
-OCOCH(OH)C6H5 6
The newly synthesized µ-oxy, derivatives of tricyclohexyl an- timony (v) are white or off white crystalline solid with sharp melting points and are soluble in organic solvents. The com- pounds are listed with their physical properties in Table-1. The elemental analysis was found satisfactory and within permis- sible limits. The data obtained are summarized in Table (3-5). The molar conductance values of the compound in acetonitrile (10−3 solution) were found in the appropriate value to show their non-conducting behavior in solution. The molecular weight and thus Vant Hoff factor 'i' determined cryoscopically in nitro benzene confirmed their monomeric nature.
The infrared absorption for all the µ-oxy bis [tricyclo- hexylantimony (v)] derivatives were recorded in the range
4000-400 cm−1 using KBr pellets on FT IR spectrophotometer (Shimadzu 8201/PC). The Characteristic absorption frequen- cies are listed in Table (3). Absorption associated with various internal modes of vibrations of µ-oxy bis [tricyclohexyl anti- mony (v)] derivatives have been identified and indicate the nature of bonding. Bond due to antimony – oxygen – antimo- ny (Sb – O – Sb) bond is at almost similar position in the range
740-725 cm−1 as strong to very strong band and come in the range of earlier published data [20, 25-26, 27]. The position of asymmetric and symmetric modes and separation (∆v) be- tween them provides a method of assessing carboxylate coor- dination modes. The non-conducting the monomeric nature and the absence of band at 1556, 1413 and 650 cm−1 due to car- boxylate ion [28-31] in the IR spectra further rule out the pos- sibility of an ionic structure. The ν (OH) for the compounds (1-
6, 9-10) was observed in the range 3465-3240 cm−1 as a weak or
medium hand. In case of mandelic acid derivatives (7, 8)
where the cyclic product was obtained due to participation of
hydrogen of carboxylate group and hydroxyl group, ν(OH)
was not recorded.
The 1H NMR spectra of some derivatives of µ-oxy bis [tricy- clohexyl antimony (v)] were recorded on Bruker DRX-300 (300
Figure 1
In other word this is due to diaxial, axial, equatorial and
diequitorial coupling of protons of rigid cyclohexyl system.
All the axial and equatorial protons of cyclohexyl ring give
signals at different δ-values. The signals for proton (H1) bond-
ed to ipso carbon of cyclohexyl ring appear in the range δ(3.60
– 3.80) ppm. The axial proton at position – 2 (H2a) and equato-
rial proton at position – 3 experienced the same electronic en-
vironment and were found magnetically equivalent giving out
the signal at same position in the range δ(1.75 – 1.85) ppm. In
this way, the signals for cyclohexyl protons were found in
good agreement with those reported earlier [31, 32]. The α-
hydroxy acids containing α-hydrogen (2-6, 9, 10) shows the
proton signal for α-hydrogen in the range δ(4.95 – 5.15) ppm.
In the case of compounds, the methoxy proton was observed
at δ(3.78, 3.81) ppm, respectively, as singlet. The phenyl ring
protons appear in the range δ(6.85 – 7.95) ppm as multiplet,
the details are summarized in the Table (4). The three, mag-
netically non equivalent protons (H2, H3, H4) of 2-pyrazine
carboxylate were observed at δ(9.0315), 8.75(d) and 8.85(d)
ppm, respectively, the peaks of ligands are comparable with
free carboxylic acids and with those of similar organometallic
compounds reported earlier [23, 24, 28-40].
13C NMR spectra of representative compounds were recorded on 300 MHz FT NMR (Bruker DRX – 300) spectrometer operat- ing at Ω75 MHz using CDCl3 as solvent and reference (δ77.0) ppm with chemical shift being reported as δ(ppm). The peaks are compared, identified and are listed in Table-5.The chemical shift of four magnetically non-equivalent of cyclohexyl ring were observed at different δ-values in the region δ(23.03 –
56.72) ppm. The ipso-carbon (C1) was more shielded in all cases and the signal appears in the range δ(55.09 – 56.72) ppm. The chemical shift behaviour of carbon centers of phenyl ring is also dependent on the pKa values of the carboxylic acids. The signals for magnetically non-equivalent carbon centers of ligand acid appeared at different δ-value; and the signal for carboxyl (>C = O) carbon was observed in the range δ(165.1 –
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 46
ISSN 2229-5518
174.75) ppm with a shiftment towards lower field in compari- son to that of free acid which indicates the participation of carboxylate group in the formation of Sb-O-C(O) bond. In case of compounds (3, 4, 6 – 8) the chemical sift values for α-c ap- pears in the range δ(73.02 – 81.60) ppm. These values are at lower field in comparison to free acids showing the deshield- ing effect at α-carbon center due to the coordination of car- boxylate group to antimony. The 2-pyrazine carboxylate de- rivatives give the distinct signals for magnetically non- equivalent carbons. The 19F NMR spectra, in case of compound (5, 6) which contains trifluoromethyl group was also recorded and a signal appeared at δ62.9 ppm for fluorine atom. Thus, on the basis of molecular weight, conductivity, IR and NMR (1H, 13C and 19F) spectral data, the µ-oxy bis [tricyclohexyl an- timony (v)] derivatives prepared in the present investigation may be assigned a trigonal bipyramidal structure with a Sb-O- Sb linkage and on each antimony atom more electronegative group are situated at apical position (fig. 1-2). The similar structure has also been established for other µ-oxo, derivatives of antimony [9, 13, 15, 19, and 20].
Tricyclohexylantimony dichloride and µ-oxo bis [tricyclo- hexylantimony (V)] chloride were prepared by the reported method [21]. The carboxylic acids (all form Aldrich) were used in the form of their silver salts. IR spectra were recorded in solid state, using KBr pellets, on FT-IR spectrophotometer (Schimadzu 8201 PC, Shimadzu Corporation Kyoto, Japan) over the spectral range 4000-400 cm-1. 1H, 19F, and 13C NMR spectra were recorded on a Bruker spectrometer (Bruker Cor- poration, US), using TMS, CF3 COOH and CDCl3 as reference, respectively. The stoichiometry of compounds was established by elemental analysis on a semimicro scale using an elemental analyzer (Elementar Vario EL III, Carlo Erba 1108, Milan, Ita- ly). All the reactions of [(cyclo– C6 H11 )3 SbCl]2 O with silver salt of corresponding – carboxylic acids were performed under dark condition in presence of inert atmosphere. The corre- sponding silver salts of carboxylic acids were prepared from their sodium salt by reaction with silver nitrate. Tetra hydro furan (THF), was distilled purified and dried under an inert atmosphere from sodium – benzophenone. The typical exper- iments for some representative µ-oxy-bis [tricyclohexyl anti- mony (v)] carboxylates are given below and the further details for the other reactions are summarized in Table (1). The ana- lytical data are listed in Tables (2-5).
A solution of [(Cyclo – C6 H 11 )3 SbCl]2 O (0.829g, 1.0 mmol) and
silver salt of salicylic acid (0.490g, 2.0m mol) in THF (25 mL)
was stirred at room temperature for 24h. On filtration of het-
erogeneous solution containing precipitate of silver chloride, a
clear solution was obtained which was concentrated in vacuo
(2-3 mL). After the addition of n-hexane (3 mL), the solution
was allowed to stand overnight at O0C affording a white crys-
talline solid which was recrystallised from a mixture of THF
and n-Hexane (1:3). The Compound was characterized as µ-
oxy bis [tricyclohexylantimony (v)] (salicyclate)] (1). In the
same manner, 1:1 molar ratio reaction of [(Cyclo-
C6 H11 )3 SbCl]2 O (0.829g, 1.0m mol) with silver salt of salicylic
acid (0.245 g, 1.0m mol) in THF (20 mL) afforded off white crystalline compound characterized as tricyclohexyl antimony (V) salicyclate µ-oxo tricyclohexyl- antimony (v) chloride (1).
In an inert atmosphere a solution of [(Cyclo – C6 H11 )3 SbCl]2 (0.414g, 0.5m mol) and silver salt of (RS) mandelic acid (0.259g, 1.0m mol) in THF (20 mL) was stirred at room tem- perature for 24 h. The insoluble portion of heterogeneous solu- tion containing precipitate of silver chloride was filtered. The filtrate on concentration in vacuo followed by addition of pe- troleum ether (40-600C) afforded white crystalline solid. The compound was crystallized from a mixture of benzene and petroleum ether (40-600C) in the ratio (1:2) and was character- ized as cyclometalled (RS) - µ-oxy bis [tricyclohexyl antimony (V)] mandelate (8). Similarly, 1:1 molar ratio reaction [(Cyclo – C6 H11 )3 SbCl]2 O (O.82gg, 1.0m mol, with silver salt of (RS) mandelic acid (0.259 g, 1.0 mmol) in THF (20 mL) afforded the same cyclometallates (RS) - µ-oxy bis [tricyclohexyl antimony (V)] mandelate (7).
A heterogeneous solution of [(Cyclo – C6 H 11 )3 SbCl]2 O (0.415g,
0.5 m mol) and silver salt of (RS) – p – (trifluoromethyl) man-
delic acid (0.327 g, 1.0m mol) in THF (15 mL) was stirred to- gether at room temperature for 24h. The white precipitate of AgCl thus formed was filtered off. The filtrate on concentra- tion under vacuum (2-3 mL) afforded while solid after addi- tion of petroleum ether (60-800C) (2mL). The solid compound was crystallized from a mixture of THF and n-Hexane (1:3) and was characterized as µ-oxy bis [tricyclohexyl antimony (V) p-(trifluoromethyl) mandelate] (5).
In the similar fashion 1:1 molar ratio reaction of [(Cyclo – C6 H11 )3 SbCl]2 O (0.829g, 1.0m mol) and silver salt of p- (trifluoromethyl) mandelic acid (0.327g, 1.0m mol) in THF (20 mL) afforded the compound (RS) tricyclohexyl antimony (V) p-(trifluoromethyl) mandelate µ-oxo-tricyclohexylantimony (V) chloride (6).
In an oxygen and moisture tree atmosphere, a solution of
[(Cyclo-C6 H11 )3 SbCl]2 O (O.829g, 1.0m mol) with silver salt of
2-Pyrazine carboxylic acid (0.462 of, 1.0m mol) in dry acetone (20 mL) was stirred at room temperature for 12h followed by refluxion for 2h. The silver chloride thus formed was filtered off and filtrate on concentration in vacuao (2mL) was stirred at
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 47
ISSN 2229-5518
room temperature for 12h followed by addition of petroleum ether (60-800C) (3mL), yielded white crystalline/4-hexane mix-
TABLE – 1 Preparation and Properties of µ-Oxy bis [tricyclohexyl antimony (v)]
![]()
derivatives
ture (1:3) to afford µ-oxybis [tricyclohexylantimony (V) (2-
Comp. Complex [Cyclo-
Ligand No. (g) Molar Ratio/ M.P.
Colour Recrystalization
pyrazine carboxylate)] (11).
No.
C6H11)3
![]()
SbCl]2O
Solvent
(0C)
Solvent
(1) [(cyclo-C6H11)3Sb]2O(Cl)(OCOC6H4OH-o) 0.829g 1, (0.245) 1:1 /THF 142 White n-Hexane
Benzene
(2) [(cyclo-C6H11)3Sb]2O(OCOC6H4OH-o)2 0.829g 1, (0.490) 1:2 /THF 165 off white THF/n-Hexane
![]()
![]()
![]()
![]()
L (cyclo-C6H11) Sb O
![]()
![]()
(cyclo-C6H11) Sb
L
(cyclo-C6H11) (cyclo-C6H11)
(cyclo-C6H11) (cyclo-C6H11)
Where L=o-OH-C6H4COO-,(C6H5)2C(OH)COO- R' = p-CF3C6H4-, p-CH3O-C6H4- & 2pyrazine carboxylate
(3) [(cyclo-C6H11)3Sb]2O[OCOC(OH)(C6H5)2]2 0.414g 4, (0.335) 1:2 /THF 115 White THF Pet-ether
(40-600)
(4) [(cyclo-C6H11)3Sb]2O(Cl)[OCOC(OH)(C6H5)2] 0.829g 4, (0.335) 1:1/ THF 98 White THF,
n-Hexane
(5) [(cyclo-C6H11)3Sb]2O[OCOCH(OH)C6H4CF3-p]2 0.415g 2, (0.327) 1:2/ THF 122 white THF, n- Hexane
(6) [(cyclo-C6H11)3Sb]2O(Cl)[OCOCH(OH)C6H4CF3-p] 0.829g 2, (0.327) 1:1/ THF 119 White THF,
n-Hexane
(7) [(cyclo-C6H11)3Sb]2O[OCOCH(O)C6H5]** 0.829g 6, (0.259) 1:1 /THF 98 White Benzene/
Pet. ether
(40-600C)
(8) [(cyclo-C6H11)3Sb]2O[OCOCH(O)C6H5]** 0.414g 6, (0.259) 1:2/ THF 98 White Pet. Ether(40-
600C)
(9) [(cyclo-C6H11)3Sb]2O[OCOCH(OH)C6H4OCH3-p]2 0.414g 4, (0.290) 1:2 /THF 135 off white Acetone,
n-Hexane
(10) [(cyclo-C6H11)3Sb]2O(Cl)[OCOCH(OH)C6H4OCH3-p] 0.414g 4, (0.145) 1:1/ THF 85 off white Acetone/ n- hexane
(11) [(cyclo-C6H11)3Sb]2O(OCOC4N2H3)2* 0.829g 5, (0.462) 1:2/Acetone 127 white Chloroform, n-Hexane
Fig 1 Showing structure of µ-Oxo derivatives
![]()
*OCOC4N2H3 = 2-pyrazine carboxylate
** = cyclometallates
![]()
![]()
![]()
cyclo-C6H11
(cyclo-C6H11) (cyclo-C6H11)
(cyclo-C6H11)
![]()
( )
![]()
![]()
![]()
Sb O Sb
![]()
(cyclo-C6H11)
![]()
O
![]() (cyclo-C6H11)
(cyclo-C6H11)
O
![]()
C (1) C43H71ClO4Sb2 930.99 55.50 (55.47) 7.59 (7.69) -
(2) C50H76O7Sb2 1032.65 63.35 (63.38) 7.39 (7.42) -
O
(3) C64H88O7Sb2 1212.90 63.35 (63.38) 7.38 (7.31) -
Fig 2 Showing structure of µ-Oxo Cyclometallate derivatives
(4) C50H77ClO4Sb2 1021.12 58.51 (58.81) 7.52 (7.60) - (5) C54H78F6O7Sb2 1196.70 54.31 (54.20) 6.53 (6.57) - (6) C45H72ClF3O4Sb2 1013.02 53.25 (53.35) 7.25 (7.16) - (7) C44H72O4Sb2 908.56 58.25 (58.17) 7.87 (7.99) - (8) C44H72O4Sb2 1120.76 57.88 (57.87) 7.60 (7.55) - (9) C45H75ClO5Sb2 975.05 55.35 (55.43) 7.70 (7.75) -![]()
(11) C46H72N4O5Sb2 1004.61 54.98 (55.00) 7.18 (7.22) 5.57 (5.58)
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 48
ISSN 2229-5518
![]()
TABLE – 4 1H NMR Spectral Data for µ-Oxybis [tricyclohexyl antimony (v)] Derivatives in δ(ppm)
![]()
ν(OCO) ν(OH) ν(Sb-C)
ν (Sb-O-
Comp. No.
Cyclohexyl Ligand
![]()
H1 H2e H2a/H3a H3e H4e H4a H2' H3' H4' OCH3 Hα
![]()
νasym νsym ∆ν
γ-mode
(2) 3.75-3.81
(m)
2.80-2.88
(m)
1.71-1.75
(m)
1.52-1.55
(m)
1.57-1.61
(m)
1.16-1.24 7.48-7.77 6.51-6.60 7.02-7.08 - -
(m)
(3) 3.80-3.85
2.91-2.87
1.73-1.78
1.52-1.56
1.18-1.20
1.19-1.25 7.25-7.48
7.14-7.17 (m) - -
(m)
(m)
(m)
(m)
(m)
(m)
(m)
(4) 3.77-3.80
2.82-2.89
1.71-1.77
1.54-1.58
1.60-1.65
1.16-1.21 7.23-7.74
7.10-7.12 (m) - -
(m)
(5) 3.79-3.83 (m)
(m)
2.82-2.90 (m)
(m)
1.72-1.76 (m)
(m)
1.53-1.58 (m)
(m)
1.61-1.64 (m)
(m)
1.16-1.19 (m)
(m)
7.40-7.78 (m) - - 5.37 (s)
(7) &
3.64-3.69
2.70-2.81
1.70-1.76
1.55-1.62
1.64-1.68 1.13-1.20 7.34-7.59
7.13-7.18 (m) - 5.28 (s)
(8)
(m)
(m)
(m)
(m)
(m)
(m)
![]()
(9) 3.81-3.85 (m)
2.89-2.95 (m)
1.73-1.79 (m)
1.51-1.57 (m)
1.60-1.63 (m)
1.14-1.19 (m)
7.45-7.82 (m) - 3.80 (s) 5.34 (s)
![]()
Where m = multiplet; d = doublet; s = singlet
H
H1
H
H3a
![]()
O O C
HO H α
![]()
O O C
HO H α
Where, w = weak, m = medium, s = strong, vs = very strong, O = overlapped
H
eH4
H4a
eH3
H
H2a
Sb
H2e
2' 2'
3' 3'
4' 4'
OCH3
Cylohexyl group attached with antimony
Carboxylate ligand attached with antimony
![]()
TABLE – 5 13C NMR Spectral Data for µ-oxybis [tricyclohexylantimony (V)] derivatives in δ ppm
Comp. No.
Cyclohexyl Ligand
![]()
C1 C2 C3 C4 Cα > C = 0 C1' C2' C3' C4'
(2) 55.43 30.01 26.14 26.00 - 173.09 141.52 130.54,
161.48 (OH)
116.98 &
118.56
134.83
![]()
(3) 55.09 29.61 26.34 26.02 81.58 174.61 143.02 127.73 127.28 127.32 (7) & (8) 55.31 29.63 28.23 25.90 73.68 174.13 139.71 128.26 126.58 127.46 (6) 55.24 29.45 28.20 25.87 73.02 173.14 139.85 126.44 126.49 128.06 (11) 56.58 28.05 25.03 25.55 - 165.01 145.61 143.98 144.53 197.66
# 19F NMR Spectra for compound (S) gave the signal at δ62.9 ppm for − CF3 group
![]()
O O
Sb C
HO H
4 3 2
![]()
α
1'
2'
3' 4'
O
N C
2' O
3'
N
Cyclohexyl group directly attached with antimony
4'
Ca rboxylate ligands attached with antimony
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 49
ISSN 2229-5518
CONCLUSION
Thus, form the IR and NMR (1H, 19F and 13C) spectral studies aided by molecular weight and conductance measurements, it is evident that carboxylic acids hehave as monodentate except in case of mandelic acid which forms cyclometallates. Thus in these newly synthesised carboxylates, the antimony is in pen- tacoordinated state impartind trigonal-bipyramidal (TBP) structure around the antimony atom, in which electronegative groups occupy apical positions and three cyclohexyl group are situated at the equitorial positions (Fig. 1). The preffered ge- ometry of five coordinated group 15 element is TBP, which is fluxional, stereochemically non rigid, or pseudorotating ar- rangement rapidly interconverting to suare pyramidal struc- ture. This has been established in case of antimony [23]. The compounds are monomeric and non electrolyte in solution having pentacoordinate dispensation around antimony atom.
Acknowledgment
The authors are thankful to Head, Chemistry Department, University of Lucknow, Lucknow, U.P., India for providing necessary laboratory facilities and to University Grant Com- mission, New Delhi, India for providing financial assistance to Dr. (Mrs.) Kiran Singhal through a major research project vide no. 37-429/2009 SR.
REFERENCES
[1] J. L. Wardell, Arsenic, Antimony and Bismuth, In: Compre- hensive Organometallic Chemistry II, Ist Edn., G. Wilkinson, F. G. A. Stone and E. W. Abel (Edrs.), Pergamon Press, Inc., New York, 321, 1995.
[2] G. T. Morgan, E. M. G. Micklehwait and G. S. Whitty, J.
Chem., 97, 34, 1910.
[3] G. Wittig and K. Clauss, Ann. Chem., 577, 26, 1952.
[4] R. G. Goel and D. R. Ridley, Inorg. Nucl. Chem. Lett., 7, 21,
1971.
[5] R. G. Goel and H. S. Prasad, Inorg. Chem., 11, 1241, 1972.
[6] A. Ouchi, M. Nakatani, Y. Takahashi, S. Kitazima, T. Sugiha- ra, M. Matsumota, T. Uchiro, K. Kitano, K. Kawashima and H. H. Handa., Sci. Pap. 25, 73 (1975).
[7] V. V. Sharutin and V. T. Byckov, Org. Khim., 4, 1191, 1991. [8] A. Ouchi and S. State, Bull. Chem. Soc. Jpn., 61, 1806, 1988. [9] E. Maslowsky, J. Organometal. Chem., 70, 153, 1974.
[10] M. A. G. M. Tinga, M. K. Groenveld, O. S. Akkerman, F. Bick- elhaupt, W. J. J. Smeets and A. L. Spek, Rec. Trav. Chim. Pays- Bas, 110, 290, 1991.
[11] R. Ruther, F. Huber and H. Preut, J. Organometal. Chem.,
342, 185, 1988.
[12] M. Shindo and R. Okawara, J. Organometal. Chem., 5, 537,
1966.
[13] G. Ferguson and D. R. Ridley, Acta. Crystal. 29B, 2221 (1973). [14] G. G. Long, G. O. Doak and L. D. Freedman, J. Am. Chem.
Soc., 86, 209 , 1964.
[15] K Bajpai and R. C. Srivastava, Synth. React. Inorg. Met.-Org.
Chem., 15(3), 327, 1985.
[16] Prem Raj, A. K. Saxena, K. Singhal and A. Ranjan, Polyhe- dron, 4, 251, 1985.
[17] Prem Raj, R. Rastogi, K. Singhal and A. K. Saxena, Polyhedron, 5, 1581, 1986.
[18] E. R. T. Tiekink, J. Organometal Chem., 333, 199, 1987.
[19] M. N. Gibbons, A. J. Blake and D. B. Sowerby, J. Organomet-
al. Chem., 543, 217, 1997.
[20] M. N. Gibbons and D. B. Sowerby, J. Organometal. Chem.,
555, 271, 1998.
[21] S. Agnihotri, Prem Raj and K. Singhal, Synth. React. Inorg.
Met-Org Chem., 32(2), 399-417, 2002.
[22] S. Agnihotri, Prem Raj and K. Singhal, Synth. React. Inorg.
Met-Org Chem., 32(3), 399 , 2002.
[23] P. Raj, S, Agnihotri, K. Singhal, Synth. React. Inorg. Met.-Org.
Chem., 32(3), 569-581, 2002.
[24] S. Agnohotri, P. Raj, K. Singhal, Synth. React. Inorg. Met.-Org, Chem., 32 (3), 449-464, 2002.
[25] K. Singhal, R. Mishra, P. Raj, Heteroatom Chemistry,19, 7, 688-
693, 2008.
[26] R. Mishra, K. Singhal, A. Ranjan, A. K. Saxena, Heteroatom
Chemistry, 21, 3, 181-187, 2010.
[27] H. Baruki, S. J. Coles, J. F. Costello and M. B. Hursthouse, J.
Organometal Chem. 622, 265 , 2001.
[28] H. Baruki, S. J. Coles, J. F. Costello, T. Gelbrich and M. B.
Hursthouse, J. Chem. Soc, Dalton Trans., 2319 , 2000. [29] W. J. Geary, Coord. Chem. Rev., 7(1), 81, 1971.
[30] G. O. Doak, G. G. Long and L. D. Freedman, J. Organometal.
Chem., 4, 82, 1965.
[31] R. G. Goel and D. R. Ridley, J. Organometal. Chem., 182, 201
1979.
[32] R. M. Wing and K. P. Callahan, Inorg. Chem., 8, 871 (1969). [33] G. B. Deacon, F. Huber and R. J. Phillips, Inorg. Chem. Acta.,
104, 41, 1985.
[34] J. S. Li, Y. Q. Ma, L. Yu, J. R. Cui and R Q. Wang, Synth. Re-
act. Inorg. Met. Org. Chem., 32(3), 583, 2002.
[35] K. Nakamoto, Inorganic Compounds, In: Infrared and Raman
Spectra of Inorganic and Coordination Compounds, 3rd Edn., John Willey and Sons, Inc. New York, 71, 1986.
[36] S. J. Blunden, B. N. Patel, P. J. Smith and B. Sugavanam, Appl.
Organometal. Chem., 1, 241, 1987.
[37] F. Huber, M. Vornefeld, H. Preut, E. V. Angerer and G. Ruisi,
Appl. Organometal. Chem., 6, 597, 1992.
[38] S. K. Shukla, A. Ranjan and A. K. Saxena, J. Fluorine Chem.,
113, 155, 2002.
[39] A. K. Saxena, A. Ranjan, K. Mishra and P. Venkataramani, Ind. J. Chem., 37A, 736 1998.
[40] M. Yanga, T. Miura, K. Endo., H. Nakahana and M. Takeda, Bull Chem. Soc., Jpn., 59(10), 3085, 1986.
[41] J. Holecek, K Handler, A. Lycka, T. K. Chattopadhyay, B.
Mazee and A. K. Kumar, Collec. Czech. Chem. Commun., 51,
1100, 1986.
[42] S. Ali, F. Ahmad, M. Mazhar, A. Munir and M. T. Masood, Synth. React. Inorg. Met.-Org. Chem., 32(2), 357, 2002.
[43] H. W. Acton and L. E. Napier, Ind. Med. Gaz., 64, 147, 1929. [44] S. K. Rathi, P.K. Pandhi, N. Khanna and P. Chopra, Ind. J.
Dermat. Venerology and Leprology, 69, 392, 2003.
[45] F. Huber, M. Vornfeld, H. Prent, E. V. Angerer and G. Ruisi, Appl. Organo. Metal. Chem., 6, 597, 1992.
[46] V. K. Avasthi, S. N. Bhattacharya and M. Verma, J. Ind.
Chem. Soc., 52, 264, 1982.
[47] C. Silvestru, C. Socaciu, A. Bara and I. Haiduc, Anti. Cancer
Res., 10, 803, 1990.
[48] C. Socaciu, A. Bara, C. Silvestru and I. Haiduc, In Vivo, 5 425 (1991), Anticancer Res., 11, 1651, 1991.
[49] C. Socaciu, I. Pasca, A. Bara, C. Silvestru and I. Haiduc, Metal
Based Drugs 1, 291, 1994.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 50
ISSN 2229-5518
[50] S. Z. Hu, L. D. Tu, Y. Q. Hang and Z. X. Li, Inorg. Chim. Acta,
232, 161, 1995.
[51] J. S. Li, Y. Q. Ma, J. R. Cui and R. Wang., Appl. Organometal
Chem., 15, 639, 2001.
[52] J. S. Li, Y. Q. Ma, L. Yu, J. R. Cui, Synth. React. Inorg. Met.
Org. Chem., 32 (3), 583, 2002.
[53] L. Yu., Y. Q. Ma, G. C. Wang and J. S. Li, Heteroatom Chem.,
15(1), 32, 2004.
[54] P. J. Craig, Applied Organometallic Chemistry, V-26, I-10, 2012.
[55] M. M. Amini, H. R. Khavasi and E. R. Tiekink, Applied Organo- metallic Chemistry, V-26, I-9, P.471-477, 2012.
[56] S. J. Higgins, R. J. Nicholas, S. Martin, P. Cea, Organometallics, V-
30, I-1, p. 7-12, 2011.
[57] F. R. Hartley, L. D. Freedman, G. O. Doak, “The use of Organoan- timony and Organobismuth Compounds in Organic Synthesis”, John Wiley & Sons Ltd., 2010.
[58] P. Sharma, D. Perez, A. Cabreza, Acta Pharmacologica Sinica, 29,
881-890, 2008.
IJSER © 2013 http://www.ijser.org