International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 23
ISSN 2229-5518
Synthesis and Characterisation of Some New
Tris (α-Naphthyl) Bismuth (V) Carboxylates; (α-C10H7)3BiX2
Kiran Singhal*, Dharmendra K. Srivastava, Prem Raj
Abstract—A series of tris (α-naphthyl) bismuth (V) carboxylates (α-C H ) Bi(OCOR) where -OCOR = p-trifluoromethyl mandelate, p- methoxy mandelate, salicylate, 2pyrazine carboxylate, mandelate have been synthesised and characterised by solid state infrared, 1H and
13C NMR spectroscopy, molecular weight and conductance measurements. The newly synthesized derivatives are monomeric in benzene and non ionic in acetonitrile. In the solid state a penta coordinate dispensation around bismuth having monodentate OCOR group indicated on the basis of spectroscopic and physic chemical datas.
Keywords— Tris (α-naphthyl) bismuth carboxylates, IR, 1H NMR, 13CNMR spectra, monomeric, non ionic
—————————— ——————————
Despite a considerable amount of work done on the synthesis, reactivity and characterization on organoantimony and or- ganoarsenic carboxylates, corresponding studies on organo- bismuth compounds are strikingly lacking. It is probably due to the difficulties encountered in the preparation of parent organobismuth compounds which requires strict anhydrous
conditions otherwise tend to decompose or are susceptible to
halocarboxylates. All the reactions were carried out in an oxy- gen free atmosphere in anhydrous organic solvent at room temperature and under dark condition. These reactions may be represented in the sense of equation shown below (Eq. 1-2):
THF, Benzene
hydrolysis. However, the introduction of bulkier cyclohexyl and naphthyl group on to the bismuth atom render hydrolytic and thermal stability and therefore easy to handle and charac- terize. Apart from this sterically congested organobismuth compounds have been little studied vis-à-vis aryl and alkyl![]()
(αC10H7)3BiBr2 + 2ML (αC10H7)3BiBr2 + ML
![]()
Benzene
(α-C10H7)3BiL2 + 2MBr (1)
(α-C10H7)3Bi(Br)L + MBr (2)
![]()
N COO-
group substituted organobismuth compounds and also hold good promise to be biologically potential due to the presence of bismuth-carbon bond which is biodegradable and bismuth as such is non toxic [1-14].
The present investigation focused on the synthesis of hither to
unreported tris(α-naphthyl) bismuth (V) carboxylate with a
view to establish the nature and mode of bonding of carbox-
ylate group. The newly synthesized compounds were charac-
terised by solid state infrared (IR), 1H, 13C NMR spectra. Solu-
tion phase studies complementary these have been carried out
to establish their behavour in solution [15, 16].
The 1:1 and 1:2 molar ratio reactions of triorganobismuth (V) dibromide with silver salt of corresponding carboxylic acid offered a series of trioganobismuth (V) dicarboxylates and –
————————————————
• *Corrseponding author is Dr. (Mrs.) Kiran Singhal, Associate Professor, Department of Chemistry, University of Lucknow, Lucknow, U.P., India-
226007 and is Ph. D. supervisor of Mr. D. K. Srivastava.
• Email: singhal.kiran@gmail.com, Ph: +91-9415159894
• Contract Grant Sponsor: University Grants Commission, New Delhi, India Vide Letter No. 37-429/2009 SR.
Dr. Prem Raj is Senior Professor Chemistry Departrment, Lucknow
University, Lucknow, U. P., India.
[Where ML; M = Ag; L = o-OHC6H4COO, RCH(OH)COO ( R = C6H5-, p-CH3O-C6H4-, P-CF3-C6H4-, ![]()
N
In case of pyrazine carboxylic acid (9, 10), and all the α- hydroxy carboxylic acids moieties both 1:2 and 1:1 molar ratio reactions proceed smoothly and afforded different products (Eq 1-2). The mandelic acid and its derivatives forms, cyclome- tallates with triphenylbismuth (V) dichloride but the same was not observed here. There observation indicates that sterically, hindered α-naphthyl group prohibit the cyclisation. The new- ly synthesized compounds do not decompose on storage for several days and are quite stable toward air and moisture. The yields of products were nearly quantitative except for the loss- es during workup process. The complexes are freely soluble in most of the organic solvent. They are off while crystalline sol- id, and have sharp melting points. The complexes have been characterized by elemental analysis, IR and NMR (1H, 13C) techniques. They have been found non-electrolyte in acetoni- trile (10-3M) and shown monomeric constitution in freezing benzene [35].
Infrared spectra of all the compounds (1-10) were recorded in the 4000-400 cm−1 region on FT IR spectrophotometer (Shi- madzu 8201 PC) IR absorptions corresponding to α-naphthyl group bound to bismuth are in dose conformity with the re- ported values of analogous tris (α-naphthyl) bismuth and – antimony [17,18]. The diagnostic IR absorption bonds of the
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 24
ISSN 2229-5518
newly synthesized compounds have been identified and are listed in Table. The infrared spectra of all the compounds show almost identical absorption due to α-naphthyl group. The Bi-C stretching frequency corresponding to y-mode was observed in the range 430-410 cm−1 as a medium to weak band [19, 20, 21]. The Bismuth – halogen, stretching frequency in spectra of the halocarboxylates are not observed in the range of 4000-400 cm−1. IR spectra provider a method of assessing corboxylates co-ordination made from the position and sepa- ration (∆V = ν asym − ν sym ) between the asymmetric and symmet- ric OCO stretching modes [22-24]. In the JR spectra of these compounds the carboxylate bonds are observed in the charac- teristic region: ν asym (OCO) between 1606 and 1630 cm−1, ν sym (OCO) between 1385 and 1310 cm−1. On the basis of ∆v (OCO) values (335 – 275 cm−1) these compounds belong to the same class [21]: there are no interaction between the carbonyl oxy- gen atom of the carboxylate group and the bismuth atom, which indicate the presence of monodentate 'ester type' – OCO group.
1H NMR Spectra of the representative compounds were rec- orded on Brucker DRX – 300 (300 MHz FT NMR) spectrometer using CDCl3 as solvent with chemical shift being reported as δ(ppm) from tetramethyl silane (TMS) as reference. The inte- gration of the spectra is in good agreement with the expected values in the complex molecule. The proton Magnetic reso- nance spectra of newly synthesized compounds are listed in Table-4. The chemical shifts of the Protons of naphthyl group appear in the range δ(7.50 – 8.20) ppm in the form of multiplet. The appearance of signals of protons of naphthyl group as multiplet is due to low to long-range coupling. In this way, the signals for naphthyl protons were found in good agreement with those reported earlier [10, 11]. The α-hydroxy acids con- taining α-hydrogen (comp. no. 3-8), shows the proton signal for α-hydrogen in the range δ (4.96 – 5.13) ppm. In case of compound (7, 8) containing the methoxy protons was ob- served at δ(3.78, 3.81) ppm respectively as singlet. The phenyl ring proton appears in the range δ(6.55 – 7.95) ppm as multi- plet, the details are summarized in the table 4. The three mag- netically non equivalent protons (H2 , H3 , H 4 ) of 2-pyrazine carboxylate (9, 10) were observed at δ 9.05 (s), 8.78(d) and 8.85 (d) ppm, respectively. The peaks of ligands (corboxylates) with free carboxylic acid and with those of similar organometallic compounds reported earlier [16, 31].
The 13C NMR spectra of some representative compounds were recorded on 300 MHz FT NMR (Bruker DRX – 300) spectrome- ter operating at ~75 MHz using CDCl3 as solvent and refer- ence (δ 77.0 ppm) with chemical shifts being reported as δ (ppm). The peaks are compared identified and are listed in Table-5. The chemical shift of some magnetically non- equivalent carbon of naphthyl ring were observed at different δ-values in the C1 – 129, C2 , - C3 , - C6 , - C7 = 126.5; C4 , C5 , C8 , at 128.5 ppm and C 9 – C10 at 133.9 and 133.7 ppm respectively. The position of the carbon centre of carboxylate group in all complexes shifts toward lower field due to deshielding as
compared to the free acid moieties, which indicates the partic- ipation of carboxylate group in co-ordination to Bismuth [32]. The positions of the carbon of ligands are recognized and have the chemical shift values in range. In case of all the sterically congested Organo-bismuth(V) carboxylates, the chemical shift of ligands shows effective difference from the chemical shift of carbon of free acid for carbon centers C1 and C12 , but other carbons are almost on affected. The effect of sterically congest- ed naphthyl group on chemical shift of Carbon canters of lig- and acid is remarkable when compared to those of triphenyl bismuth (V) carboxylates [16, 31]. The presence of all peaks of carbons of naphthyl group and ligand acid on one hand and the change in chemical shift of different carbon centers of lig- and acid compared to those of free acid on the other hand con- firms the formation of Bi-O-C(O) bond. Thus on the basis of spectral data in combination with molecular weight and con- ductance data, it may be concluded that the newly synthesized compounds possess a monomeric, bismuth. The three α- naphthyl group (s) occupy the equatorial position and the electronegative group (s) ligand or chlorine occupies axial po- sitions (Fig. 1) [31-42].
The preparation of (α−C10 H7 )3 BiBr2 was prepared by reported method [5]. The carboxylic acids were used in the form of sil- ver salts which were obtained by reacting sodium salt of cor- responding acid with silver nitrate. Tetrahydrofuran (THF) was distilled under an atmosphere of nitrogen from sodium- benzophenone. Special precautions were taken to exclude moisture and oxygen. The reactions of silver salts were done under dark condition to avoid decomposition. Typical Exam- ples of (α-C10 H7 )3 BiBr2 and its mono and di carboxylate de- rivative (α-C10 H7 )2 BiBr (OCOR'), (α-C10 H7 )3 Bi (OCOR) 2 re- spectively are described below in detail and the conditions for the other reaction are summarized in Table-1. Analytical data are given in Table-2 to 5.
The heterogeneous solution of tris (α-naphthyl) Bismuth (v) dibromide (0.75g, 1.0 m mol) and silver salt of salicylic acid (0.49 g, 2.0 mmol) in THF (25 ml) was stirred at room tempera- ture for 24 h. The AgCl thus formed wais filtered off, the fil- trate was concentrated in vacuo (3-4 ml) followed by addition of n-hexane (5 ml) afforded white crystalline solid which was characterized as tris (α-naphthyl) bismuth (V) disalicyclate (2). Similarly 1:1 molar ratio reaction of tris (α-naphthyl) bismuth (V) dibromide (0.75g, 1.0 mmol) with silver salt of salicylic acid (0.245 g, 1.0 m mol) in THF (25 ml) afforded white crystal- line solid characterized as tris (α-naphthyl) bismuth (V) Bro- mo Salicyclate (2).
In an atmosphere of dry nitrogen a solution of tris (α- naphthyl) bismuth (V) dibromide (0.750 g, 1.0 m mol) and sil- ver salt of (RS) mandelic acid (0.518 g, 2.0m mol) in THF (25 ml) was stirred at room temperature for 24h. The heterogene-
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 25
ISSN 2229-5518
ous solution containing precipitate of AgCl was filtered. The filtrate on concentration in vacuo followed by addition of Pe- troleum ether (60-800C) afforded white crystalline solid. The compound was crystallized from a mixture of THF and petro- leum ether (60-800C) in the ratio 1:4 and was characterized as (Rs) – tris (α-naphthyl) bismuth (v) di mandelate (3). In the same manner, 1:1 molar ratio reaction of tris (α-naphthyl) bis- muth (V) dibromide (0.75 g, 1.0 m mol) with silver salt of (Rs)
– mandelic acid (0.259 g, 1.0 m mol) in THF (20 ml) offered the
![]()
Table – 1 Preparation and Properties of Tris (α-naphthyl) bismuth (V) halo carboxylates and dicarboxylates
white crystalline compound characterized as (RS) tris (α-
naphthyl) bismuth (V) (bromo) (mandelate) (4).
Comp. No.
Complex Organometallic
Halides
![]()
(g)
Ligand
(g)
Molar Ratio/ Solvent mL
M.P. C (0C)
In an inert atmosphere, a heterogeneous solution of tris (α- naphthyl) bismuth (V) dibromide (0.75g, 1.0 mmol) and silver salt of 2-pyrazine carboxylic acid (0.462 g, 2.0m mol) in dry benzene (20 ml) was stirred at room temperature for 24h. The silver nitrate, thus formed was filtered off completely and sol- vent was evaporated under vacuum to afford off white crystal- line solid. The compound was recrystalized from a mixture of benzene and petroleum ether (60-800C) in the ratio 1:3 and was characterized as tris (α-naphthyl) bismuth (V) di (2-pyrazine carboxylate) (9). In the same manner, 1:1 molar ratio reaction of tris (α-naphthyl) bismuth (V) dibromide (0.75g, 1.0 m mol) with silver salt of 2-pyrazine carboxylic acid (0.388 g, 1.0 m mol) in THF (15 ml) afforded the off white crystalline com- pound characterized as tris (α-naphthyl) bismuth (V) (Bromo) (2-pyrazine carboxylate) (10).
L/Br
1 (α-C10H7)3Bi(Br)(OCOC6H4OH-o) (α-C10H7)3Bi Br2
(0.75)
2 (α-C10H7)3Bi(OCOC6H4OH-o)2 (α-C10H7)3 BiBr2
(0.75)
3 (α-C10H7)3Bi[OCOCH(OH)C6H5]2 (α-C10H7)3BiBr2
(0.75)
4 (α-C10H7)3Bi(Br)[OCOCH(OH)C6H5] (α-C10H7)3BiBr2
(0.75)
6 (α-C10H7)3Bi(Br)[OCOCH(OH)C6H4CF3-p] (α-C10H7)3BiBr2
(0.75)
7 (α-C10H7)3Bi[OCOCH(OH)C6H4OCH3-p]2 (α-C10H7)3BiBr2
(0.75)
8 (α-C10H7)3Bi(Br)[OCOCH(OH)C6H4OCH3-p] (α-C10H7)3BiBr2
(0.75)
9 (α-C10H7)3Bi[OCOC4N2-2]2* (α-C10H7)3BiBr2
(0.75)
10 (α-C10H7)3Bi(Br)[OCOC4N2-2]2* (α-C10H7)3BiBr2
(0.75)
![]()
*OCOC4N2-2 is 2-pyrazine carboxylate
[o-OHC6H4COOAg] (0.245)
[o-OHC6H4COOAg] (0.490) [C6H5CH(OH)COOAg] (0.518)
[C6H5CH(OH)COOAg] (0.250)
[p-CF3C6H4CH(OH)COOAg] (0.327)
[p-CH3OC6H4CH(OH)COOAg] (0.578)
[p-CH3OC6H4CH(OH)COOAg] (0.462)
[C4N2COOAg] (0.462)
[C4N2COOAg] (0.388)
1:1 THF (25)
1:2 THF (25)
1:2 THF (25)
1:2 THF (20)
1:1 THF (20)
1:2 THF (25)
1:2 THF (15)
1:2 THF (25)
1:2 THF (15)
105 W
138 w
118 w
85 w
136 w
148 w
129 w
110 o
98 o
![]()
![]()
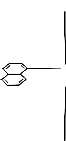
Bi
L
Fig:1Showing TBPY arrangement of
tris(α-naphthyl)bismuth(V) halo-carboxylates and dicarboxylates;
where L = o-OHC6H4COO,C6H5CH(OH)COO, p-CF3-C6H4CH(OH)COO,
p-MeOC6H4CH(OH)-COO,2-pyrazine carboxylate
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 26
ISSN 2229-5518
The replacement of phenyl group (C 6 H5 -) by bulkier organic moiety (i.e. α-naphthyl) enhances the hydrolytic stability thus it can also expect that it can affect the biological activity. The carboxylates group acts as monodentate group. The com- pounds are monomeric and non electrolyte in solution having pentacoordinate dispensation around bismuth atom.
The authors are thankful to Head, Chemistry Department, University of Lucknow, Lucknow, U.P., India for providing necessary laboratory facilities and to University Grant Com- mission, New Delhi, India for providing financial assistance to Dr. (Mrs.) Kiran Singhal through a major research project vide no. 37-429/2009 SR.
[1] K. Singhal, R. Rastogi, P. Raj, Indial Journal of Chemistry, v-
26A, pp. 146-150, 1987.
[2] K. Singhal, P. Raj, Synth. React. Inorg. Met-Org. Chem., 23(6), pp. 1011-1020, 1993.
[3] F. Jee, A. K. Aggarwal, P. Raj, Proc. Indian natn. Sci. Acad, 59, A, 3, pp 309-313, 1993.
[4] G. B. Deacon, W. Roy, J. and J. M. Pfieffer, Aust. J. Chem., 37,
pp 527-535, 1984.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 7, July-2013 27
ISSN 2229-5518
[5] T. Hasan, P. K. Singh, R. Mishra, P. Raj, N. Misra, Pramana
Journal of Physics, v-68, 5, pp. 875-880, 2007.
[6] Th. Klapotke, Journal of Organometallic Chemistry, 331, pp.
299-307, 1987.
[7] S. Agnihotri, P. Raj, K. Singhal, Synth. React. Inorg. Met-Org.
Chem., 32 (3), pp. 449-464, 2002.
[8] P. Raj, S. Agnihotri, K. Singhal, Synth. React. Inorg. Met-Org.
Chem., 32 (3), pp. 569-581, 2002.
[9] S. Agnihotri, P. Raj, K. Singhal, Synth. React. Inorg. Met-Org.
Chem., 32 (2), pp. 399-417, 2002.
[10] K. Singhal, R. Mishra, O. Prakash, Asian Journal of Chemistry, v-19, 1, pp 288-296, 2007.
[11] K. Singhal, P. Raj, R. Mishra, Heteroatom Chemistry, 19 (7), PP.
688-693, 2008.
[12] K. Singhal, R. Mishra, O. Prakash, A. Jour. Chem., 19 (1), pp.
288-296, 2007.
[13] K. Singhal, P. Raj, R. Mishara, Heteroatom Chemistry, O. Pra- kash, Synth. React. Inorg. Met-Org. Chem., 35 (10), pp. 889-893,
2005.
[14] R. Mishra, K. Singhal, A. Ranjan, A. K. Saxene, P. Raj, Heteroa- tom Chemistry, 21, 3, 2010.
[15] H. Baruki, S.J. Coles, J.F. Castello and M.B. Hursthouse, J
.Organometal. Chem, 622,265, 2001.
[16] R.M.Silverstein G. C. Bassler and T. C. Morrill, In: Spectroscop- ic identification of organic compounds, John Wiley and Sons. Inc.: New York, 4th Edn, 95, 1981.
[17] S. N. Bhattacharya and I. Hussain, J. Ind. Chem. Soc., 4, 1119,
1981.
[18] G. O. Doak, C. G. Long and L.D.F reedman, J. Organometal.
Chem, 4, 82, 1965.
[19] J. S. Li, G. Q. Huang, Y. T. Wei, C. H. Xiang, D. Q. Zhu and Q.
L. Xie, Appl. Organometal Chem., 12, 31. 1998.
[20] J. S. Li, Y. Q. Ma, J. R. Cui and R. Q. Wang, Appl. Organometal
Chem., 15, 639, 2001.
[21] K. Singhal, R. Rastogi and P. Raj, Ind. J. Chem., 26A, 146, 1987. [22] M. N. Gibbons and D. B. Sowerby, J. Organometal Chem., 555,
271, 1998.
[23] J. S. Li, Y. Q. Ma, J. R. Cui and R. Q. Wang, Synth. React. Inorg.
Met.-Org. Chem., 32(3), 583, 2002.
[24] J. S. Thayer and R. West, Adv. Organometal. Chem., 5, 169,
1967.
[25] A. H. Norbury and A. I. P. Sinha, J. Chem. Soc., A, 1598, 1968. [26] R. G. Goel and D. R. Ridley, Inorg. Chem., 13, 1252, 1974.
[27] S. N. Bhattacharya, A. K. Saxena and P. Raj, Ind. J. Chem., 21A,
141, 1982.
[28] T. N. Srivastava and S. N. Bhattacharya, J. Inorg. Nucl. Chem.,
28, 2445, 1966.
[29] R. G. Goel, Canadian J. Chem., 47, 4607 1969.
[30] H. Barucki, S. J. Coles, J. F. Costello, T. Gelbrich and M. B.
Hursthouse, J. Chem. Soc., Dalton. Trans., 2319, 2000.
[31] J. Holecek, K. Handler, A. Lycka, T. K. Chattopadhyaya, B.
Majee and A. Kumar, Collec. Czech. Chem. Commun., 51, 1100,
1986.
[32] V. K. Jain, R, Bohra and R. C. Mehrotra, J. Ind. Chem. Soc., LVII, 408, 1980.
[33] G. Alonzo, N. Bertazzi and M. Consiglio, Inorg. Chim. Acta. 85,
35, 1984.
[34] C. Silvestru, C. Socaciu, A. Bara and I. Haiduc, Anticancer Res.,
10, 803, 1990.
[35] W. J. Geary, Coord. Chem. Rev., 7, 81, 1971.
IJSER © 2013 http://www.ijser.org