International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1221
ISSN 2229-5518
Huntington’s Disease
Sukanya Rajan
Abstract: Huntington's Disease is a destructive, progressive and dynamic neurological disorder coming about fundamentally, from degeneration of nerve cells profound in the central brain. It was discovered by George Huntington in 1872, after whom it has been named. It is inherited as an autosomal dominant condition.The side effects of HD generally show up between the ages of 30 - 50 years, but in rare cases they have been observed to be initiated in ages younger than five and as old as seventy.The course of Huntington's Disease can be anywhere in the range of ten to thirty years from onset to death. It influences all races and ethnicities and happens with the same recurrence in both males and females. In March 1993, researchers discovered that HD is brought on by a mutation in a gene found on chromosome 4. At present, the disease is incurable, however dynamic advancement has been made as specialists investigate the condition and make considerable headway. Albeit not very many instances of HD are because of new mutations, everybody who has the HD gene will inevitably inherit the disease unless he or she passes away from some other cause before the signs show up. Current exploration and research revolves around the transgenic mouse model created in Berlin at the Max Planck Institute. There is practically no choice of residential care to those suffering from HD, particularly in the later phases of the ailment. In any case, the quest for a cure is the need of the hour, and families and groups are joining together to promote better treatment and understanding for those with the fatal disease.This paper concentrates on a more intensive take at the Huntington gene and evaluating the most pervasive disclosures in the journey for discovering a conceivable cure.
(HD – Huntington’s disease)
KEYWORDS : Huntington’s disease, neurological disorder, chromosome 4, gene mutation, autosomal dominant disorder, cures, brain cells, inheritance.
—————————— ——————————
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1222
ISSN 2229-5518
1 INTRODUCTION
George Huntington described it as "coming on gradually but surely, increasing by degrees, and often occupying years in its development until the hapless sufferer is but a quivering wreck of his former self".
The HTT gene is found on the short arm of chromosome 4 at
4p16.3, containing an arrangement of three DNA bases— cytosine-adenine-guanine (CAG)—recurring various times (i.e. ... CAGCAGCAG ...), called as a trinucleotide repeat. CAG is the 3-letter codon for the amino acid glutamine, so an arrangement of them brings about the creation of a polyglutamine tract, and the repeated part of the gene is called the PolyQ region. By and large, individuals having less than 36 repeating glutamines in the polyQ region produce the cytoplasmic protein Huntingtin. [1] (Walker FO,
2007).
However, a grouping of 36 or more glutamines brings about the generation of a protein which has distinctive characteristics. For the most part, the quantity of CAG repeats is identified with how much this process is influenced and variation is credited to environment and other modifying genes that alter HD mechanism.
Trinucleotide CAG repeats in excess of 28 are unstable while replicating and this instability builds with the number of repeats present. This typically prompts new extensions as generations pass (dynamic mutations) as opposed to recreating an accurate duplicate of the trinucleotide repeat. This causes the quantity of repeats to change in progressive generations, such that an unaffected parent with a "moderate" number of repeats (28–35), or "decreased penetrance" [2] ( "Unified Huntington's Disease Rating Scale (UHDRS)"UHDRS and Database,2009) may pass on a duplicate of the quality with an expand in the number of repeats that creates completely penetrant Hd. Such expands in the quantity of repeats in successive generations is known as genetic anticipation. Instability is more noteworthy in spermatogenesis than oogenesis; as paternally inherited alleles are susceptible to having an increase in length.
Medical diagnosis of the onset of HD could be carried out
after appearance of physical side effects particular to the disease. [3] (Walker FO, 2007). Genetic testing might be utilized to affirm a physical finding if there is no family history of HD. Indeed before the onset of manifestations,
genetic testing can affirm if an individual or embryo conveys
an extended duplicate of the trinucleotide repeat in the HTT quality that causes the ailment. Genetic councelling has also come into existence to give guidance to affected people on coping with personal and professional lives post-diagnosis, and on the ramifications of an affirmed finding. Notwithstanding the accessibility of presymptomatic testing, just 5% of those at danger of inheriting HD decide to get tested for it.
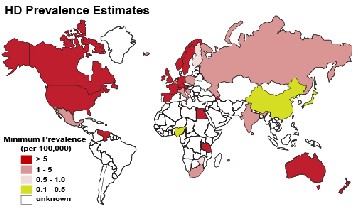
Fig. 1. Huntington’s disease prevalence worldwide
2 CAUSES AND PATHOLOGY
2.1 Causes
Huntington's disease is brought about by an inherited defect in a single gene. A guardian parent with a damaged Huntington gene could pass along the defective duplicate of the gene or the sound duplicate. Every child in the family, in this way, has a 50 percent probability of inheriting the causative gene.
The ordinary duplicate of the gene delivers a protein called huntingtin, however the broken quality is bigger than typical and produces a bigger type of huntingtin.
Cells in parts of the brain – particularly, the basal ganglia and parts of the cortex – are exceptionally sensitive to the impacts of the unusual huntingtin.
It is brought about by an irregular extension of polyq (>35) space in the N-end of the Huntingtin protein (Htt). [4] (Zuccato C, Valenza M, Cattaneo E (2010)
The overall predominance of HD is 5–10 cases for every
100,000 persons.
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1223
ISSN 2229-5518
2.2 Pathology
One of the histopathological marker of HD is intranuclear aggregates made by the polyQ-changed Htt protein. These insoluble structures trap critical proteins and also can physically impair cell activity and change for example the ubiquitin-proteasome degradation process. By and large these deformities at lead to the neurodegeneration happening essentially in the striatum, and later including other territories at later phases of the disease.
Despite the fact that it is acertained that the disease onset happens as a result of stretched polyQ, different regions of Htt could additionally impact the pathogenicity of polyQ- hhtt. In reality, the initial 17 amino acids (N17 area) and the proline-rich region (PRR district), both adjoining the polyQ stretch were found to impact aggregation. [5] (Arribat et al.
2014)
Different theories have developed: loss of trophic support, glutamate excitotoxicity, mitochondrial dysfunction, and autophagy. Each of these speculations is legitimate as for the capacity of mutant HTT (mhtt), yet no single hypothesis brings together the distinct pathology of HD. [6] (Crane et al, 2014)
Histopathologically, HD is portrayed by sudden death of medium-sized (essentially GABA-ergic) striatal neurons, yet vast interneurons are for the most part saved.
[7] (J. P. G. Vonsattel, R. H. Myers, T. J. Stevens et al, 1985.) Notwithstanding the advancement of responsive gliosis
inside the striatum, loss of the grey matter is far reaching
and brings about the compensatory extension of the lateral brain ventricles. Because of the general atrophy, the brain weight can diminish by 40% [8] (J. F. Gusella, 2001)
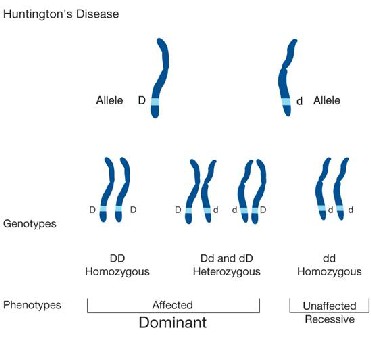
Fig. 2. Genomic view of Huntington’s disease inheritance.
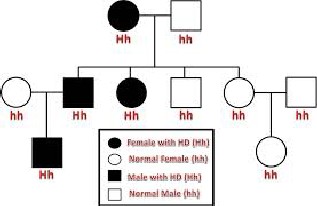
Fig. 3. Pedigree chart of Huntington’s disease inheritance
3 SIDE EFFECTS
The three most significant behavioral issues in Huntington 's sickness originate from the uncontrollable movements called "chorea," dementia, and the modified perception of the world. As the illness advances, the intensity of the side
effects expand and could be divided into three stages.
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1224
ISSN 2229-5518
3.1 Primary stage

The primary stage is described by subtle and slight uncontrolled movements, including staggering and clumsiness, concentration deficit and fleeting memory passes, and depression and mood changes.
3.2 Secondary stage
Amidst the illness' course, patients may require support with daily activities because walking, speech and swallowing abilities deteriorate. Involuntary movements tend to be more conspicuous and may be observable to the onlooker. In the later stages, patients with HD need full-time care, and
regularly families select to place them in nursing homes.
3.3 Tertiary stage
Cognitive capabilities are continuously impaired. Especially influenced are executive capacities which incorporate planning, cognitive adaptability,abstract thinking,rule acquisition, initiation of appropriate actions, and inhibition of inappropriate actions. Reported neuropsychiatric signs are depression, anxiety, a decreased presentation of feelings (blunted effect), egocentrism, aggression, and compulsive behaviour, the last of which can result in or intensify addictions, including liquor addiction, gambling, and hypersexuality. difficulties in perceiving other individuals' negative expressions have also been noted.
[9] (Montoya A, Price BH, Menear M, Lepage M (2006)
Self-destructive considerations and suicide attempts are more common than in the normal people.
3.4 Physiological and anatomical changes
Mutant Huntingtin is communicated all through the body and connected with irregularities in peripheral tissues that are directly caused by such expression outside the brain. These anomalies incorporate muscle destruction, cardiovascular failure, impeded glucose tolerance, weight reduction, osteoporosis, and testicular atrophy. [10] (van der Burg JM, Björkqvist M, Brundin P (2009).
Patients might never again have the capacity to talk, rigidity might now be to a greater extent an issue than involuntary movements. Feeding tubes regularly must be utilized in light of the fact that patients can no more swallow. On the other hand, even as the ailment advances, individuals with the disease quite often recognize their families, are mindful of the circumstances, and can comprehend. Death normally
occurs due to pneumonia, malnutrition, or heart failure.
Fig. 4 Physiological symptoms of Huntington’s disease
4 RESEARCH
AND ADVANCEMENTS
1.in HD mouse models and human patients, the presence of obvious mhtt aggregates called inclusion bodies (Ibs) relates with the oncoming of behavioral shortfalls [11] (Davies et al.,
1997). IB shaping is confined anatomically notwithstanding
omnipresent outflow of mhtt. Inclusion body creation demonstrates a mismatching between the generation and leeway of aggregation prone protein. The process of IB shaping in HD is indistinct, yet the length of the poly Q repeat area corresponds with the quantity of Ibs in diseased brains [12] (vonsattel et al.,1985; Becher et al.,1998)
Notwithstanding amassed mhtt,ibs contain ubiquitin, molecular chaperones, and proteasome subunits
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1225
ISSN 2229-5518
,recommending that cells have lacking ability to clear misfolded mhtt [13] (Sieradzan et al.,1999; Stenoien et al.,1999; Waelter et al.,2001; Mitra and finkbeiner,2008).
Additional proof proposes that cells can break down Ibs much after they are formed: [14] (Yamamoto et al, 2000) created an inducible HD mouse display in which they ended mhtt creation after Ibs and behavioral shortages arose. Turning off mhtt generation brought about Ibs to vanish and turned around the behavioral deficiencies.
2. The presence of mhtt in neural cells is by all account not the only instrument, which is included in HD pathogenesis. Metabolic and mitochondrial dysfunctions, oxidative and nitrative anxiety, furthermore apoptosis assume a vital part too [15] (S. E. Browne and m. F. Beal, 2006)
3. it is generally realized that the neurodegenerative process of HD phenotype is a chronic procedure, morphologically described by the dynamic degeneration of neurons, basically in the striatum, however steadily influencing just about all parts of the cerebrum. This results in a diminishment of grey matter and brain atrophy with compensatory broadening of the horizontal cerebrum ventricles. By and by, additionally
as the second segment of the cerebrum parenchyma, the glial
cells play a key part in this procedure. In this study, histopathological gimmicks, huge for adjustments in human brains experienced HD and brains of tghd51 CAG rats, were compared. The critical distinction is essentially in the force of neuronal degeneration which, indeed in the most established rats, does not achieve the seriousness of the harm trademark for cutting edge phases of HD in people. In tghd51 rats, a lot of striatal neurons show just continuous decline in their size and don't completely worsen also vanish until the death of creature (at around 2 years of age). In this way, likewise concomitant reactive astrogliosis is definitely not broad in old tghd rats, dissimilar to the progressed phases of HD in humans. on the other hand, striatal decay (the decrease of grey matter) creates steadily, and it is recognizable from 12 months of age in tghd rats, showed fundamentally by compensatory expansion of horizontal brain laterals. The explanation behind striatal shrinkage is the same in rats and people; it begins from the slow degeneration of neurons coming about in rarefaction of the neuropil incorporating the changes in the thickness and character of neurotransmitters.
4. Transplantation of stem cells for the treatment of
Huntington's illness (HD) accumulated much consideration
preceding the turn of the century. Many studies utilizing mesenchymal stem cells (Mscs) have shown that these cells have gigantic potential in HD. Preferences of utilizing Mscs for cell treatments incorporate their simplicity of isolation, rapid propagation in culture, and favorable immunomodulatory profiles. Be that as it may, the absence of steady neuronal differentiation of transplanted Mscs has constrained their helpful viability to abating the movement of HD-like side effects in animal models of HD.
The general triumphs of utilizing methodologies for reducing neuropathological and behavioral deficits in rodent models of HD have respectable potential for clinical utilization. In any case, the decision of what sort of hereditarily changed undifferentiated cell to use for transplantation is reliant on the phase of HD and whether
the deciding objective is safeguarding endogenous neurons
in ahead of schedule stage HD, or supplanting the lost neurons in late-arrange HD. [16] (Crane et al,2014)
5. In 2008, the USDA approved tetrabenazine to treat the motor indications of HD, while anti psychotics and antidepressants have been endorsed to treat other developmental, cognitive and emotional disturbances. [17] [ Shoulson, I.; Young, A.b. 2011]. On the other hand, these medications are just palliative and not generally viable. Subsequently, it is important to discover medications that
can mitigate the manifestations and delay the progress of the ailment.
It has been entrenched that diminishments in pro cell survival neurotrophic elements (Ntfs, for example, brain derived neurotrophic element (BDNF), is significantly diminished in the HD cerebrum. The BDNF protein, delivered in the cerebral cortex and transported through corticostriatal tracts to Msns, is essential for MSN survival.
To further comprehend the significance of BDNF in the striatum of the HD brain, transgenic HD mice were crossed with heterozygote BDNF knockout mice, which quickened the progress of the disease phenotype [18]( Altar, C.A.; Cai, N.; Bliven, T. et al, 1997)
[19] (Canals, J.M.; Pineda, J.R.; Torres-Peraza, et al. 2004) On the other hand, crossing HD transgenic mice with mice
that overexpress the BDNF gene postponed progression of
the disease and secured against neuropathological dysfunction.
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1226
ISSN 2229-5518
Despite the fact that, it ought to be noted that when the HD- BDNF transgenic cross arrived at 16 months, the animals started to show epileptic-like seizure movement, in all likelihood in light of the fact that the level of BDNF was not endured.
[20] ( Xie, Y.; Hayden, M.R.; Xu, B. 2010)
Conveyance of restoratively huge measures of Ntfs is testing. Although repeated peripheral administrations of fibroblast development consider 2 into mouse models of HD have demonstrated some adequacy
[21] (Jin, K.; LaFevre-Bernt, M.; Sun, Y.; Chen, S.et al 2005) conveyance of numerous different Ntfs, including BDNF, into the HD brain remains a test, because of the way that
numerous Ntfs are substantial, polarized proteins that do not readily cross the blood-brain-barrier, relegating the delivery of NTFs directly into the central nervous system.
[22]. (Pardridge, W.M. 2007)
[23] (Dragatsis, I.; Levine, M.S.; Zeitlin, S. 2000)
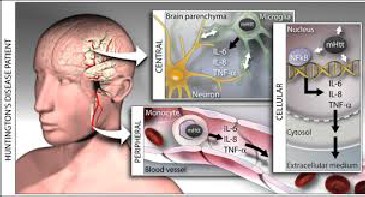
Fig. 5 Internal changes during Huntington’s disease
5 TREATMENT
5.1 Gene therapy
Gene therapy is a guaranteeing new avenue for the
treatment of HD. Because of the relative straightforwardness of single mutation on a single gene, numerous techniques to lessen the measure of the transformed protein have
advanced as of late. Be that as it may, the essential properties
of the HTT protein and the ubiquitous expression of HTT in all cells has given difficulties to gene treatments. A real snag is that HTT is essential for ordinary development. This has been indicated by Duyao and associates who found that Htt null mice die at embryonic day 7.5 [42].
[24] (Duyao, M.P.; Auerbach, A.B.; Ryan, A.et al 1995)
5.2 Macroautophagy (henceforth alluded to as autophagy) It sequesters perpetual proteins, organelles, or parasites
inside double membrane autophagosomes [25] (Rubinsztein et al., 2007), which combine with lysosomes to degrade the sequestered substance. Misfolded and accumulated mhtt may weaken autophagy instigation in striatal neurons. Case in point, mhtt expres sion lessens the representation of Omi/Htra2 in civilized striatal neurons and in human HD striatum [26] (Inagaki et al., 2008). Omi/Htra2, a mitochondrial chaperone and protease [27](Clausen et al.,
2002), manages autophagy and mitophagy [28] (Li et al.,
2010; Cilenti et al., 2014). In this manner, diminished outflow of autophagy- related proteins and decreased impelling of autophagy may make striatal neurons more defenseless against mhtt.
5.3 Proteostasis pathway
The striatum gets critical dopaminergic and excitatory glutamatergic inputs from the substantia nigra and cerebral cortex,respectively.excito toxicity caused by glutamatergic indicating through N-methyl-D-aspartate receptors (nmdars)may help striatal-particular degeneration in Hd [29](levineetal.,1999; Zeron et al.,2002). This expanded affectability to NMDAR initiation might additionally influence striatal proteostasis instruments. The territorial selectivity of IB shaping shows that distinctions in striatal proteostasis limit are in charge of the specific degenera tion of Msns. recent confirmation backs this case, exhibiting that parts of autophagy, the UPS, and chaperone systems are communicated or managed contrastingly in striatal neurons than in other brain areas. Accordingly, focusing on proteostasis pathway particularly in the striatum may uncover new treatments for HD.
One method to lessen levels of the transformed protein is the utilization of proteins that work intracellularly to tie to target proteins, or intrabodies. At the point when the intrabodies
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1227
ISSN 2229-5518
bind to mhtt, the complex is ubiquitinated, stamping it for degradation and removal. Two specific intrabodies, Happ1 and Em48, brought into the striatum by means of viral vectors can lessen a portion of the neuropathological and behavioral side effects in HD mice
[30] Qin, Z.-H.; Wang, Y.; Sapp, E.; Cuiffo, B.; Wanker, E.; Hayden, M.R.; Kegel, K.B.
[31] (Southwell, A.L.; Bugg, C.W.; Kaltenbach, L.S.; Dunn, et al 2011)
On the other hand, the capability to successfully convey intrabodies, specificity to mhtt, and to counteract
cytoplasmic protein misfolding, and in addition to push long
term manifestation decrease are still indistinct and oblige further examination.
[32] (Butler, D.C.; McLear, J.A.; Messer, 2012)
5.4 RNA interference (Rnai).
This engineering utilizes the endogenous RNA-induced silencing complex, which binds the anti sense micro-RNA, or interfering RNA, to mRna which then either severs or wrecks the double stranded RNA. The most regularly utilized Rnai techniques have been virally brought into the brain through AAV vectors. In healthy non-human primates Aavs conveying micro Rna against wild-type HTT in the rhesus macaque demonstrated a 45% diminishment in HTT inside the putamen, with no antagonistic events noted.
[33] (Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L. et al
2005)
[34] (Rodriguez-Lebron, E.; Denovan-Wright, E.M et al 2005) [35] (DiFiglia, M.; Sena-Esteves, M.; Chase, K. et al 2007)
[36] (McBride, J.L.; Pitzer, M.R.; Boudreau, R.L.; Dufour, B. et al)
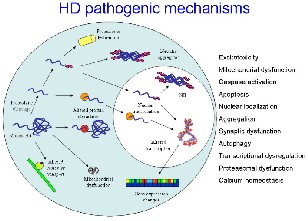
Fig. 6 Pathogenic mechanisms
6 RECENT RESEARCH
6.1 Mesenchymal stem cells
In the previous year, scientists have published on the use of stem cells to model Huntington disease (HD) and on investigations of stem cell use as a treatment for HD. Sadan and colleagues1 prompted mesenchymal cells into neurotrophic factor–secreting (NTF) cells, then transplanted the NTF cells into rats that had striatal injuries affected by quinolinic acid (QA). The NTF cells, whether inferred from a HD patient or a control, survived transplantation and kept up a Ntf secreting phenotype prompting enhanced striatal volume and behavioral phenotypes.
6.2 Embryonic stem cells
Ma and colleagues2 demonstrated that human embryonic stem cells could be steered into an enriched population of GABA medium spiny neurons. At the point when GABA neurons were embedded into the striatum of a QA lesioned mouse model, they kept up usefulness, prompting phenotypic imporovements in the mice.
6.3 Antisense oligonucleotides
Kordasiewicz and colleagues7 utilized transiently administered antisense oligonucleotides (Asos), intended to suppress the expression of huntingtin, in HD mouse models and in non-human primates. In mouse models, Asos were directed into the lateral ventricle and prompted particular knockdown of the mhtt transgene and some phenotypic inversion.
The knockdown was supported in both animal models for
up to three months post treatment, recommending that Asos might give a clinically applicable system for fighting HD.
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1228
ISSN 2229-5518
6.4 Population studies
Ji and colleagues8 led a population based study evaluating the occurrence of disease in Swedish patients with HD and other neurological illnesses. The standardized frequency of cancer was essentially lower in patients with polyglutamine illness. The biggest contrast (0.47) was connected with HD gene carriers. Polyglutamine disease appears to be defensive against benevolent and metastatic cancer.
6.5 Drugs
Armstrong and Miyasaki9 saw the information accessible for current pharmacological choices for treating chorea. Their analysis demonstrated that tetrabenazine, the main medication affirmed by the FDA particularly for the treatment of chorea, remains the best performing medication at a measurement of 100 mg/day. Amantadine and rizuole give unassuming profit. It is essential to have numerous successful medications accessible for HD patients because of tetrabenazine's potential unfavorable impacts and the necessary close observation of patients who get the medication.
6.6 Gene silencing
It offers numerous potential profits. To start with, it does not oblige agents to focus on the exact mechanism by which mhtt causes disease. Likewise, with gene silencing, the lethal impacts of mhtt would be countered by the disruption of intracellular mhtt generation. At last, it would remove any toxicity connected with mutant mrna. The utilization of
antisense oligonucleotides (Asos) is a promising approach to gene silencing . Asos are little single stranded bits of DNA that bind via complementary base-pairing to the
intracellular mrna transcript .In HD, Asos prevent the
transcription of mhtt. Asos have been found to diminish various distinctive mhtt-related anomalies in animal models of HD.
A recent study demonstrated that infusion of Asos targetting mhtt into the brains of mouse models of HD could reduce motor side effects, prevent brain loss and expand survival rates. The profits of the ASO treatment persevered after the creation of mhtt had come back to pretreatment levels. The study additionally found that implantation of Asos into the cerebrospinal liquid conveyed the Asos to the brain and brought down mhtt mrna levels in most brain regions in
non-human primates. This strategy for conveyance could be more secure for human patients than immediate intracerebral injection and may influence a more extensive range of brain tissues. This second point is noteworthy on
the grounds that numerous research groups have demonstrated that HD neuronal pathology is not constrained to the striatum.
[37] Lise Munsie HD Insights, Vol. 3
7 CASE STUDY
Headed by Prof Sarah Tabrizi of University College London, the TRACK-HD study was intended to run like a mock drug trial. Individuals carrying the HD mutation would be examined for a characterized time of time (36 months), utilizing an extensive array of measurements including brain scans, specialized motor measurements and examination by
a doctor.
7.1 What happened?
In a fourth progressive paper published in top journal Lancet Neurology, the TRACK-HD group has recently reported its final information, describing what they saw in individuals carrying the mutation following 3 years of observation. This timing is paramount, in light of the fact that its a reasonable time span for a true therapeutic trial. It gives us a chance to answer the inquiry, 'in the event that we had a viable treatment, might we be able to test it in Huntington's disease mutation carriers in 3 years?'
There is a straightforward, confident, message that
originates from this study, and that will be that we now have better methods for doing clinical trials in HD. We know which particular tests are most sensitive to change at distinctive phases of the illness. As an outcome of this, we know what number of people we would need to certainly
see those progressions as a major aspect of any trial of a
treatment in HD patients.
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1229
ISSN 2229-5518
7.2 How'd they do it?
TRACK-HD included yearly follow ups of groups of individuals who've inherited the Huntington's disease mutation. Utilizing well established scientific computations that assist foresee when somebody with the transformation will have indications of HD, individuals without manifestations of HD were isolated into two groups: the individuals who are assessed to be near, or a long way from, infection onset.
The group additionally emulated a gathering of patients in the early phases of HD and, for comparison, a control populace who did not carry the HD mutation. Huge numbers of the control group are relatives of the HD mutation carriers.
"One hopeful message from this study is that individuals who've inherited the mutation that causes HD appear to have the capacity to adapt to it for a long while" Of the 366 people enlisted, 298 finished the 36-month catch up. Of course, numerous members that dropped out were in the more developed phases of HD.
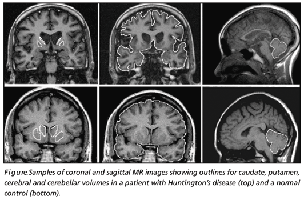
Fig. 8. Comparison of normal and affected brain
7.3 What’d they find?
the main aim of the TRACK-HD study was to determine which measurements best predict the onset of HD, and track its course after appearance of symptoms. So what did the team observe for each of the groups in the study?
First, sensitive MRI brain scans, that accurately measured the shape and size of people’s brain, could measure differences amongst every group in the study. Even the people predicted to be far from onset had specific regions of brain change during the 3-year duration of the study. Hopefully, all new studies of therapies in HD will include brain scans, so scientists can see whether this loss of brain tissue is prevented.
In the group of participants predicted to be far from onset, there was very little change in behavioral or other clinical measures during the 3-year follow up. These people seem to be coping fairly well with the changes in their brains observed by scanning.
However, over 36 months, participants predicted to be close to onset behaved rather differently. They started to show changes in a number of clinical tests, including a range of motor and memory tasks. As in the group predicted to be farther from onset, these behavioural changes were
accompanied by changes in brain that reveal shrinkage.
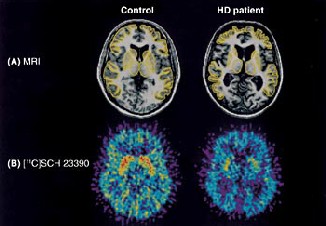
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1230
ISSN 2229-5518
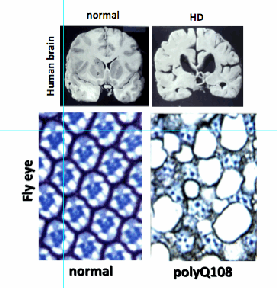
Fig. 9 Comparison of normal and affected brain
(Microscopic)
Over the 3-year duration of the study, some of the participants who hadn’t been diagnosed with Huntington’s disease at the beginning of the study have now developed symptoms of the disease. This enabled the scientists to try and figure out which measurements predicted the transition from ‘pre-manifest’ to ‘manifest’.
Several behaviors were useful in predicting onset of disease symptoms, including motor tasks such as finger tapping. Consistent with the idea that people with Huntington’s disease have a hard time with empathy and emotional regulation, people who developed disease also demonstrated problems on an emotion recognition task.
ACKNOWLEDGMENTS
The author would like to thank Amity University Noida, India for the opportunity to carry forward this paper.
CONCLUSION
Undoubtedly, Huntington’s disease is an extremely severe condition which renders a sufferer, as well as his family and friends disheartened and feeling helpless. As we know, all sufferers inevitably die of the disease sooner or later. This makes one think that some things are beyond human control, even In today’s technologically advanced era.
It is fortunate that many possible treatments are being investigated, and research is going on to find a cure of seemingly unpreventable diseases like Huntington’s. With the coming of stem cell therapy, gene therapy and many other newer forms of treatment, we can certainly hope for more progress being made in the future.
Like other polyglutamic diseases, Huntington is not a result of one’s environment, lifestyle or choices. Call it fate, or a work of nature. Sincere hopes that we come across a potent cure soon enough, to ensure people’s belief in science over destiny.
REFERENCES
[1] (Walker FO, 2007).
[2] ( "Unified Huntington's Disease Rating Scale
(UHDRS)"UHDRS and Database,2009) [3] (Walker FO, 2007).
[4] (Zuccato C, Valenza M, Cattaneo E (2010)
[5] (Arribat et al. 2014) [6] (Crane et al, 2014)
[7] (J. P. G. Vonsattel, R. H. Myers, T. J. Stevens et al, 1985.) [8] (J. F. Gusella, 2001)
[9] (Montoya A, Price BH, Menear M, Lepage M (2006)
[10] (van der Burg JM, Björkqvist M, Brundin P (2009).
IJSER © 2014 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014 1231
ISSN 2229-5518
[11] (Davies et al., 1997).
[12] (vonsattel et al.,1985; Becher et al.,1998)
[13] (Sieradzan et al.,1999; Stenoien et al.,1999; Waelter et al.,2001; Mitra and finkbeiner,2008).
[14] (Yamamoto et al, 2000)
[15] (S. E. Browne and m. F. Beal, 2006) [16] (Crane et al,2014)
[17] [ Shoulson, I.; Young, A.b. 2011].
[18]( Altar, C.A.; Cai, N.; Bliven, T. et al, 1997)
[19] (Canals, J.M.; Pineda, J.R.; Torres-Peraza, et al. 2004) [20] ( Xie, Y.; Hayden, M.R.; Xu, B. 2010)
[21] (Jin, K.; LaFevre-Bernt, M.; Sun, Y.; Chen, S.et al 2005) [22]. (Pardridge, W.M. 2007)
[23] (Dragatsis, I.; Levine, M.S.; Zeitlin, S. 2000)
[24] (Duyao, M.P.; Auerbach, A.B.; Ryan, A.et al 1995) [25] (Rubinsztein et al., 2007)
[26] (Inagaki et al., 2008).
[27](Clausen et al., 2002)
[28] (Li et al., 2010; Cilenti et al., 2014). [29](levineetal.,1999; Zeron et al.,2002).
[30] Qin, Z.-H.; Wang, Y.; Sapp, E.; Cuiffo, B.; Wanker, E.; Hayden, M.R.; Kegel, K.B.
[31] (Southwell, A.L.; Bugg, C.W.; Kaltenbach, L.S.; Dunn, et al 2011)
[32] (Butler, D.C.; McLear, J.A.; Messer, 2012)
[33] (Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L. et al
2005)
[34] (Rodriguez-Lebron, E.; Denovan-Wright, E.M et al 2005) [35] (DiFiglia, M.; Sena-Esteves, M.; Chase, K. et al 2007)
[36] (McBride, J.L.; Pitzer, M.R.; Boudreau, R.L.; Dufour, B. et al)
[37] Lise Munsie HD Insights, Vol. 3
IJSER © 2014 http://www.ijser.org