International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 806
ISSN 2229-5518
Pallavi Sahare1*, Archana Moon2 and GB Shinde3
1, 2, 3 Department of Biochemistry, RTM Nagpur University, Nagpur-440033
Corresponding author: pallavi.sahare@gmail.com
Ph.No. +91-8806218869
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 807
ISSN 2229-5518
Bacteria can be distinguished from other eukaryotic organisms due to their small size (0.2 – 10µm), they lack internal organelles, they have cell wall and they divide through binary fission. Also they lack introns, are not capable of endo/exocytosis and have single stranded circular DNA [1]. All bacteria share a common structure i.e. they possess
1. Slime: This is the extracellular material made up of polysaccharides loosely associated with the bacteria that helps them in colonisation on smooth surfaces.
2. Capsule: This polysaccharide outer coating of the bacterial surface often plays a role in preventing phagocytosis of the bacteria.
3. Cell wall: Provides bacteria its shape, form and rigidity. The cell wall is made up of the peptidoglycan that consists of alternating units of N-acetyl glucosamine (NAG) and N- acetyl muramic acid (NAM) that are cross-linked by a peptide bridge.
4. Cytoplasmic membrane: The phospholipid bilayer
5. Flagella: These offer the capacity for locomotion to the bacteria.
6. Pili: These structures project from the cell surface enabling bacteria to adhere to host tissue surfaces.
The difference between Gram positive and negative is due to a much larger peptidoglycan in Gram positives. As a result, the iodine and crystal violet precipitate in the thickened cell wall and are not eluted by alcohol in contrast to the Gram negatives where the crystal violet is readily eluted from the bacteria. As a result, bacteria can be distinguished based on their morphology and staining properties. Some bacteria such as mycobacteria (the causative agent of tuberculosis) are not reliably stained due to the large lipid content of the peptidoglycan. Alternative staining techniques (Kinyoun or Acid fast stain) are therefore used to detect these bacterial species [3].
The characteristic difference between Gram positive and negative bacteria is the presence of the amount of peptidoglycan. The bacterial peptidoglycan is rigid but flexible macromolecule that surrounds and protects the bacterial cell. Peptidoglycan serves a structural role in the bacterial cell wall, giving structural strength, as well as counteracting the osmotic pressure of the cytoplasm. Peptidoglycan is also involved in binary fission during bacterial cell reproduction. It is made up of carbohydrate backbone of altering units of N-acetyl muramic acid and N-acetyl glucosamine; these are cross linked with short peptides [4]. The peptidoglycan biosynthesis takes place in cytoplasm with the synthesis of muramyl pentapeptide precursor containing terminal D-ala D-ala. L-Alanine is converted to D-alanine by racemase, with subsequent assembly of D- alanyl-D-alanine by D-Ala-D-Ala ligase. In the cytoplasm, the muramyl pentapeptide precursor is anchored via a water-soluble UDP- glucosamine moiety. In the second phase of peptidoglycan construction, the muramyl pentapeptide N-acetylglucosamine is transferred to undecaprenyl phosphate with the release of UMP to form a Lipid I intermediate. An additional glycosylation step completes the peptidoglycan unit, which is then transported via its C55 lipid tail to the external periplasmic surface of the membrane, where the peptidyglycan unit becomes integrated into the cell wall matrix. Several transpeptidases and transglycosylases connect the newly formed
peptidoglycan structures to the cell wall peptidoglycan matrix.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 808
ISSN 2229-5518
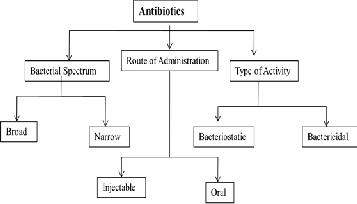
To treat various infections arising due to Gram positive, Gram negative and miscellaneous bacteria many antibiotics are used currently. The antibiotics (Greek word, anti=against and bios=life) also called as antibacterials are the types of medications that destroy/slow down the bacterial growth. The antibiotics are classified into different categories based on bacterial spectrum, site of action, chemical structure and type of activity.
The current antibacterial therapies cover a wide array of targets and can be categorized as follows:
The broad spectrum antibiotics [Carbapenems, cephalosporins, β-lactams, etc] affect a wide range of bacteria while the narrow spectrum antibiotics [Penicillin G, macrolidesphosphomycin, etc] target specific types of bacteria such as Gram positive or negative. The bacteriostatic antibiotics [examples: tetracycline, sulphonamides, spectinomycin, trimethoprim, chloramphenicol, macrolides, lincosamides] inhibit the growth of bacteria by interfering with the proteins, DNA or other cellular metabolism while the bactericidal antibiotics [examples: β-lactam antibiotics, vancomycin, aminoglycosidic antiotics like Amikacin, Arbekacin, Gentamycin, Kanamycin, Neomycin, Streptomycin] kill
bacteria.
‘Bacteriostatic’ means an agent that prevents bacterial growth or keeps them in stationary phase of growth. The bacteriostatic activity can be defined as the ratio of MBC (Minimum Bactericidal Concentration) to MIC (Minimum Inhibitory Concentration).Bacteriostatic drugs predominantly inhibit ribosome function, targeting both the 30S (tetracycline family and aminocyclitol family) and 50S (macrolide family and chloramphenicol) ribosomal subunits [5, 6, 7, 8, 9].
Tetracycline enters the cell by either passive diffusion or by energy dependent transport system. Once inside the cell, tetracycline inhibits protein synthesis by reversibly binding 30S ribosome and inhibits binding of aminoacyl-t-RNA to the acceptor site on the 70S ribosome. The protein synthesis is ultimately terminated leading to a bacteriostatic effect [10].
Spectinomycin reversibly interferes with mRNA interaction with the 30S ribosome. It inhibits translocation of the peptidyl tRNA from the A site to P site. It is structurally similar to aminoglycosides but does not cause misreading of mRNA. Aminoglycosides have many mechanisms viz; it interferes with the proofreading process which leads to increased rates of errors in protein synthesis which can give premature termination, it inhibits ribosomal translocation or it may disrupt bacterial cell membrane integrity [11].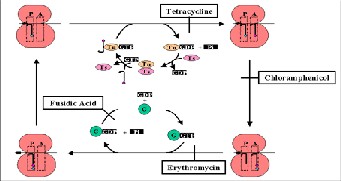
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 809
ISSN 2229-5518
Chloramphenicol, lincomycin and clindamycin bind to the
50S ribosome and inhibit peptidyl transferase activity. Chloramphenicol inhibits the peptidyl transferase thereby preventing protein chain elongation [12]. Lincomycin acts by binding with the 50S subunit of the bacterial ribosome where it prevents the binding of aminoacyl RNA to the messenger ribosome complex by inhibition of peptidyl transferase. Ultimately, bacterial protein synthesis is inhibited [13].
The macrolides inhibit translocation of the peptidyl tRNA from the A to the P site on the ribosome by binding to the 50S ribosomal 23S RNA [14]. These are generally considered to be bacteriostatic, they may be bactericidal at higher doses. Fusidic acid inhibits bacterial replication and does not kill the bacteria. It binds to elongation factor G (EF-G) and inhibits release of EF-G from the EF- G/GDP complex [15].
The bactericidals have different cellular targets. The β- lactam antibiotics target peptidoglycan biosynthesis, aminoglycosides target ribosomes, fluoroquinolones target topoisomerase. In addition to this, the bactericidals induce the formation of reactive oxygen species.
The beta lactam antibiotics inhibit the synthesis of peptidoglycan layer of the bacterial cell wall. The final step in the synthesis of peptidoglycan i.e. transpeptidation is catalyzed by DD-transpeptidase (these are Penicillin Binding proteins, PBP). These are analogous to D-Ala D-ala of the terminal peptidoglycan. The structural similarity between the beta lactam and D-ala D-ala facilitates their binding to the active site of PBP that leads to the cross-linking of
nascent peptidoglycan and hence disrupting the bacterial cell wall [16].
been initiated and induce misreading of the mRNA. These antimicrobials bind to DNA-dependent RNA polymerase and inhibit initiation of RNA synthesis. Quinolones like nalidixic acid, ciprofloxacin, oxolinic acid bind to the subunit A of DNA gyrase (topoisomerase) and prevent supercoiling of DNA, thereby inhibiting DNA synthesis [15].
The Fluroquinolones block the DNA replication pathway by binding to the A-subunit of the DNA gyrase enzyme. The DNA gyrase is a topoisomerase II enzyme that unwinds the DNA during replication. This would unable the bacteria not only from replicating the DNA, but also from protein synthesis [17].
The bactericidal antibiotics initiate Reactive Oxygen Species (ROS) formation. The molecular mechanisms include its binding to their target, disrupting the normal cellular metabolism including tri- carboxylic acid (TCA) cycle. This depletes intracellular NADH pairs with the increased production of reactive oxygen species ROS
(peroxide and superoxide). These ROS interact with Fe2+ to generate
highly toxic oxygen radicals which react with DNA and proteins, thereby, resulting in the death of bacteria [18].
Inhibitors of cell wall synthesis [β-lactam antibiotics, Glycopeptides
(vancomycin, teicoplanin), Fosfomycins]
Inhibitors of protein synthesis [Aminoglycosides(Gentamycin, Tobramycin, Amikacin, Streptomycin, Kanamycin, Netilmicin), MLSK (Macrolides, Lincosamides, Streptogramins, Ketolides), Tetracyclines (Tetracyclin, Doxycycline, Minocycline), Glycylcyclines (Tigecycline), Phenicols (Chloramphenicol), Oxazolidinnes (Linezolid), Ansamycins(Rifampin)]
Inhibitors of membrane function [Polymyxins]
Folate pathway inhibitors [Sulfonamides, Trimethoprim]
Aminoglycosides follow another mode of action; it irreversibly binds to the 30S ribosome and freezes the 30S initiation
complex (30S-mRNA-tRNA), so that no further initiation can occur. The
Inhibitors of nuc leic ac id synthes is [Quinolones, Furanes]
Structure:
aminoglycosides also slow down protein synthesis that has already
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 810
ISSN 2229-5518
Penicillins: They possess the β-lactam ring, example Ampicillin, methicillin, oxacillin etc.
Cephalosporins: These contain β-lactam ring structure. (They differ from penicillins due to the presence of a 6 – member β-lactam ring. The other difference is the existence of a functional group (R) at position 3 of the fused ring system). Examples are cefoxitin, cephamyxin, cefotaxime

Fluroquinolones: These are synthetic antibacterial agents. Tetracyclins: They are derived from Streptomyces and have a four ringed structure
Macrolides: These antibiotics are derived from Streptomyces and thus named since they possess macrocyclic lactone in its structure. Examples are Erythromycin, Azithromycin and Clarithyromycin.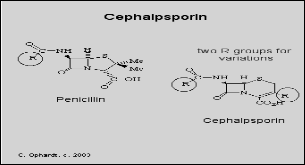
The state when bacteria show resistance to more than one antibiotic is called as Multidrug Resistance (MDR). Indiscriminate and
inappropriate use of antibiotics leads to mechanisms to evolve into a MDR strain: enzymatic deactivation of antibiotics (β-lactamase emergence of MDR strains which get modified with mutations to prevent antibiotic access or its action on the target bacteria. The bacteria utilize either of the following mentioned production), decreased cell wall permeability for antibiotics, efflux mechanism to remove antibiotic, altered target sites for antibiotic action (by mutation).
An ability of bacteria to resist the effects of multiple antibiotics to which they were previously sensitive convert them to MDR strain. Bacteria get mutated/adapted that reduces or eliminates the effectiveness of antibiotic and continue to survive without harm. There are many research papers that have identified different bacterial species which have shown resistance to multiple drugs. The commonest of them are: Acinetobacter baumannii [21,22,23]; Staphylococcus aureus [24,25,26]; Enterococcus species [27,28, 29]; Clostridium difficile [30]; Haemophilus influenzae [31]; Klebsiella pneumoniae [32,33,34]; Streptococcus pneumoniae [35,36]; Mycobacterium tuberculosis[37,38, 39]; Salmonella enterica [40,
41];Salmonella typhimurium [42, 43] and Streptococcus species [44].
The diagram (Fig 7) describes the various sites of action for different antibiotics [45].
Altered target
This type of resistance mechanism is shown by both Gram negative as well as positive bacteria. The mechanism of action for most of the antibacterial antibiotics involves interaction between the drug and intracellular enzyme/protein. These interactions either alter or inhibit the normal functions of the enzyme [46]. The development of drug resistance for these antibiotics requires the reduction in affinities to their enzymatic targets [47]. Altering an antibiotic’s target protein directly at the DNA level is a common mechanism of target
modification.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 811
ISSN 2229-5518
The beta lactam resistance is mediated by altered PBPs. The target for this antibiotic is the cell wall synthesizing enzymes called as penicillin binding proteins (penicillin sensitive enzyme). PBPs are present in almost all the bacteria but their number, size and affinity to beta lactam varies from species to species. The important PBP enzymes are peptidoglycan transpeptidase and carboxypeptidase [48]. PBPs are the membrane proteins with molecular weight ranging from
40,000 to 120,000 [49]. PBPs are the components of bacterial cell wall that play an important role in synthesis of peptidoglycan. PBP catalyses the final step of polymerisation (transglycosylation) and cross linking by transpeptidation of peptidoglycan [49].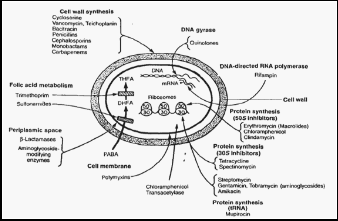
PBPs are the proteins that show an affinity towards penicillin. All β-lactam antibiotics (except tabtoxinine β-lactam) bind to PBPs. β-lactam antibiotics bind to PBP because they are structurally similar [50].
PBPs play an important role in Polymerisation of glycan strand (transglycosylation); Cross lining of glycan chains (transpeptidation); Hydrolyzation of terminal D-alanine
(carboxypeptidation) and Hydrolyzation of the peptide bond connecting the glycan strand (endopeptidation) The terminal D-Ala-D-Ala end of
peptidoglycan has structural resemblance with penicillin and hence the last stage peptidoglycan synthesizing enzymes (transpeptidases and transglycosylases) are sensitive towards penicillin that impairs their ability for peptidoglycan cross-linking [52].
Methicillin resistance in S.aureus has been associated with alterations in PBPs and found to be pH dependent. For example, the bacterial culture grown at pH 5.2 has no detectable PBP2a. Also the altered PBPs relate with the decreased susceptibility of bacteria for the antibiotic [53].
Methicillin-resistant Staphylococcus aureus is a bacterium responsible for several difficult-to-treat infections in humans. It is also known as MRSA/ORSA and it shows resistant to antibiotics called beta-lactams. These antibiotics include methicillin and other more common antibiotics such as oxacillin, penicillin, and amoxicillin. In the community, most MRSA infections are skin infections.
The United Kingdom has one of the highest levels of incidence of MRSA in Europe [54]. In 1993 there were 216 deaths where Staphylococcus infection was the final underlying cause of death. This figure rose to 546 deaths in 1998 [59]. The growing problem in the Indian scenario is that MRSA prevalence has increased from 12% in 1992 to 80.83% in 1999 [55].
S. aureus antibiotics resistant strain can cause serious health hazards via infections in community. In the 1960s, 10% of S. aureus strains produced penicillin-destroying enzymes (penicillinases); today the figure approaches 100%. Despite the introduction of methicillin in 1959 to tackle the increasing problem of penicillin resistance, it took only 3 years for MRSA to appear in 1961. Once resistance is genetically encoded it can spread rapidly within a population of bacterial species, or even to another bacterial species through transduction (the process whereby foreign DNA is introduced into another cell via a viral vector, it does not require physical contact between the cell donating the DNA and the cell receiving the DNA),
transformation (incorporation and expression of exogenous DNA from
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 812
ISSN 2229-5518
its surroundings and taken up through the cell membrane), conjugation (the transfer of genetic material between bacterial cells by direct cell- to-cell contact or by a bridge-like connection between two cells) or transposition (DNA sequence or transposable elements can change its position within the genome, sometimes creating mutations and altering the cell's genome size) [56].
MRSA is resistant to all β-lactams, penicillins, cephalosporins etc. This resistance is due to production of penicillin binding protein 2a encoded by mecA gene that has low affinity for β-lactams [57, 58]. This mecA gene can rapidly be identified with PCR that can be ultimately used for genotypic identification of methicillin resistance [59,
60].The primers that corresponded to nucleotides 181 to 200 as the sense strand and 311 to 330 as the antisense strand within mecA gene were selected for their specificities and efficiencies. The primers were named MRS1 (5'-GAAATGACTGAACGTCCGAT) and MRS2 (5'- GCGATCAATGTTACCGTAGT), respectively. This set of primers amplifies a 150-bp-long segment of the mecA gene. The ED-PCR (Enzymatic Detection of PCR) product has been explained by Ubukata et.al [61]. The transformation of methicillin susceptible S.aureus with mecA gene (chromosomally encoded PBP 2a) converts it into MRSA [62]. But the cellular level of PBP 2a does not correlate with levels of methicillin resistance. In addition to mecA, femA also functions for the methicillin resistance. femA is a chromosomally encoded 48kDa protein which affects the glycine content of peptidoglycan [63]. Also it has been shown that the presence of NaCl in the growth medium affects the phenotypic expression of methicillin resistance by affecting PBP [64,
65].
o Vancomycin resistance in enterococci
Earlier, it was believed that the mechanism behind the development of vancomycin resistance involves the thickening of the bacterial cell wall. But now it is clear that the vancomycin resistance in bacteria is due to the presence of vancomycin resistance gene-vanA. The expression of vanA is associated with alteration of vancomycin binding site in the cell
wall. Vancomycin interferes with the terminal D-Ala D-Ala of the
peptidoglycans and hence disrupts the bacterial cell wall synthesis [66,
67]
The mode of action of vancomycin in relation to peptidoglycan synthesis has been described earlier [68,69]. Vancomycin binds with high specificity to the C-terminus region of
uracil diphosphate–N-acetylmuramyl-pentapeptide containing D-Ala-D- Ala of the peptidoglycan. This prevents the addition of late precursors by transglycosylation and also prevents cross-linking by transpeptidation [69]. Vancomycin does not penetrate into the cytoplasm; therefore, interaction with its target can take place only
after translocation of the precursors to the outer surface of the membrane [68]. Mechanism of Vancomycin resistance is due to the expression of Vancomycin operon. The vanA and vanB operons are located on plasmids or in the chromosome [70], whereas the vanD [71], vanC [72], vanE [73] and vanG [74] operons have, thus far, been found only in the chromosome.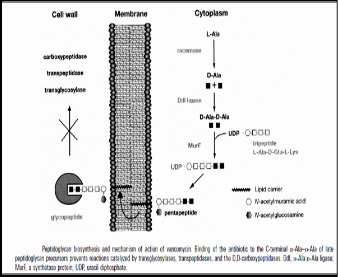
The examples of vancomycin resistance are:
• Changes in peptidoglycan layer and cell wall thickness resulting into reduced activity of vancomycin. In S. aureus, the resistance is mediated by cell wall thickening with
reduced cross linking. This traps the antibiotic before it
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 813
ISSN 2229-5518
reaches its major target, the murein monomers in the cell membrane [75].
• Changes in vancomycin precursors reduces activity of vancomycin: Enterococcus faecium and E. Faecalis
• The enzyme ((VanC, VanE, and VanG gene product) for synthesis of low-affinity precursors [C-terminal d-Ala residue of the peptidoglycan is replaced by d-lactate (d-Lac) or d- serine (d-Ser)]
• Fluoroquinolone resistance
The important mechanism behind resistance to fluroquinolones resistance involves the alterations in drug target enzymes (DNA gyrase in Gram negative and Topoisomerase IV in Gram positive bacteria) that decrease the binding affinity of the drug and alterations in the efflux pumps. Both types of alterations involve chromosomal mutations [76, 77].
Altered permeability:
This type of resistance mechanism is shown by Gram negative bacteria. This is another type of mechanism adapted by bacteria to emerge as a drug resistance strain which involves either the lack of entry of the drug inside the cell by decreased membrane permeability or greater exit via active efflux. The bacterial cell membrane is the major permeable barrier separating the extracellular environment from the intracellular environment. The fluidity of the membrane is generally balanced to include most nutrients, while excluding many toxins. Adjusting this fluidity impedes the function of the membrane. Therefore, bacteria fail to protect themselves due to change in the fluidity of its membrane, thereby leaving them unprotected. Bacteria have additional structures (Lipopolysaccharides, non specific porin channels, specific diffusion channels) [78] that surround the cytoplasmic membrane and form pores through it. The alteration of these structures to exclude antibiotics is another mechanism of antibiotic resistance.
• Outer membrane permeability
The outer membrane of bacteria acts as a selective barrier by combining the characteristics of highly hydrophobic outer membrane with porin channels for selective transports [79]. Most of the antibiotics are able to penetrate the bacterial cell envelope essentially by 2 pathways i.e. Lipid mediated diffusion pathway for hydrophobic antibiotics and Porin mediated transport for hydrophilic antibiotics. The bacteria develop antibiotic resistance by modifying any of the following
3 components viz., Permeability barrier (outer membrane), Porin channels and Altered proton motive force
The outer membrane is a bilayer of phospholipids and lipopolysaccharides (LPS) [79]. LPS is present in the outer envelope and carries net negative charge and is present mainly in Gram negative bacteria. These are responsible for impermeability of bacteria to antibiotics.
Lipid mediated diffusion pathway for hydrophobic antibiotics:
The hydrophobic antibiotics that gain entry through outer membrane bilayer include aminoglycosides (gentamycin, kanamycin), macrolides (erythromycin), rifamycin, novobiocin, fusidic acid and cationic peptides [80, 81]. Tetracylcine and quinolones use both a lipid- mediated and a porin-mediated pathway. The core-region of LPS plays a major role in providing a barrier to hydrophobic antibiotics [79].
Porin mediated transport for hydrophilic antibiotics:
Porin are hydrophilic transport channels which regulate the outer membrane permeability. There are 2 types of porins depending on their function:
a) Nonspecific diffusion porins b) Specific porins
• Efflux pumps:
The efflux pumps are present in almost all living cells and they play an important role in detoxification of antibiotics. These pumps are present on the membrane and recognize small amphiphilic
antibiotic molecules and pump them outside the cell. Bacteria use
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 814
ISSN 2229-5518
these to rid themselves of antibiotics and thus become drug resistant
[82].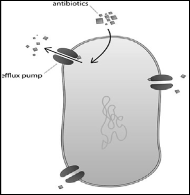
They are predominant in eukaryotes and require energy. They have been classified on the basis of their import and export activity . The import pumps are present only in prokaryotes whereas the efflux pumps are found both in prokaryotes and euaryotes 83,84[].
Production of inactivating enzymes (Gram negative/positive):
This is one of the common mechanism of developing resistance to antibiotics and shown by numerous Gram positive and Gram negative bacteria. Expression of degradative enzymes is a common mechanism by which bacteria develop resistant to antibiotics. Such enzymes chemically modify antibiotics so that they no longer function.
The genes for these degradative enzymes are obtained by acquisition
of exogenous genes or mutation of endogenous genes. The expression of these genes also governs the level of antibiotic resistance in bacteria.
o Chloramphenicol acetyltransferase (EC 2.3.1.28)
This bacterial enzyme plays a crucial role in detoxification of the antibiotic chloramphenicol, and hence plays a major role in chloramphenicol resistance.
o Aminoglycoside-modifying enzymes
Aminoglycoside are a complex family of compounds characterized by the presence of an aminocyclitol nucleus (streptamine, 2-deoxystreptamine, or streptidine) linked to amino sugars through glycosidic bonds.The aminoglyoside modifying enzymes including nucleotidyl transferase, phosphotransferase and acetyltransferase that catalyses the modification at different –OH and – NH2 groups of 2-deoxystreptamine nucleus or the sugar moieties [85].
o β-Lactamases (EC 3.5.2.6)
The main function of the enzyme β-Lactamases is to provide beta lactam antibiotic resistance to bacteria by breaking the beta lactam ring in β-lactam antibiotics (penicillin , cephamycin, carbapenems etc).
The bacteria on exposure to an antibiotic, the susceptible strains get destroyed but the fittest strain start adaptation by means of mutation and evolution. In a repetitive process and exposure to every new drug,
in due course of time, it shows resistance towards multiple antibiotics
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 815
ISSN 2229-5518
and emerges more evolved, adapted, mutated, fittest multidrug resistant bacterial strain.
1. Frank Lowy. Bacterial Classification, Structure and Function.
2. James Bartholomew and Tod Mittwer. The Gram stain.
Bacteriol Rev. 1952; 16 (1): 1-29.
3. Robin Treuer1 and Shelley E. Hayde. Acid-Fast Staining and Petroff Hausser Chamber Counting of Mycobacterial Cells in Liquid Suspension. Curr Protoc Microbiol.; PMC 2012.
4. Ghuysen. Use of bacteriolytic enzymes in determination of wall structure and their role in cell metabolism. Bacteriological reviews. 1968: 425 – 464.
5. Michael Kohanski, Daniel Dwyer, Boris Hayete, Carolyn Lawrence, James Collins. A Common Mechanism of Cellular Death Induced by Bactericidal Antibiotics. Cell 2007;
130:797-810.
6. I Chopra and M Roberts. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev.2001: 232–260
7. J Poehlsgaard and S Douthwaite. A bacterial ribosome as a target for antibiotics. Nat.Rev. Microbiol.2005:870-881.
8. T. Tenson, M. Lovmar, M. Ehrenberg. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome; J. Mol. Biol.2003:1005–1014.
9. B W eisblum and J Davies. Antibiotic inhibitors of the bacterial ribosome. Bacteriol. Rev. 1968: 493-528.
10. Schnappinger D, Hillen W . Tetracyclines: antibiotic action, uptake, and resistance mechanisms. Arch Microbiol. 1996;
165(6):359-69.
11. Shakil, Shazi, Khan, Rosina, Zarrilli, Raffaele et.al.
Aminoglycosides versus bacteria – a description of the action, resistance mechanism, and nosocomial battleground. Journal of Biomedical Science 2007;15: 5–14.
12. Bergmann, E. D., AND Sicher, S. Mode of action of chloramphenicol. Nature 1952; 170: 931-932.
IJSER © 2013 http://www.ijser.org
13. F N Chang, C J Sih, and B W eisblum. Lincomycin an inhibitor of aminoacyl sRNA binding to ribosomes. Proc Natl Acad Sci U S A. 1966; 55(2): 431–438.
14. Tenson, T.; Lovmar, M.; Ehrenberg, M. The Mechanism of Action of Macrolides, Lincosamides and Streptogramin B Reveals the Nascent Peptide Exit Path in the Ribosome. Journal of Molecular Biology 2003; 330 (5): 1005–1014
15. Dr. Gene Mayer. Bacteriology
16. Fisher, J. F.; Meroueh, S. O.; Mobashery, S. Bacterial Resistance to β-Lactam Antibiotics: Compelling Opportunism, Compelling Opportunity. Chemical Reviews
2005; 105 (2): 395–424.
17. Hooper, D. C. Mode of action of fluoroquinolones. Drugs
1999; 58(2): 6-10.
18. Gerard W right. On the road to Bacterial Cell Death. Cell
2007; 130:781-783.
19. Derek Moore. Antibiotic Classification and mechanism
20. Dr. Aisyah Saad Abdul Rahim. An Illustrated Review on Penicillin And Cephalosporin : An Instant Study Guide For Pharmacy Students. W ebmedCentral.com
21. Aharon Abbo, S. Venezia, O. Muntz, T. Krichali, Y. Igra, Y.
Carmeli. Multidrug-Resistant Acinetobacter baumannii. Emerg Infect Dis. 2005; 11(1): 22–29.
22. Federico Perez, A. Hujer, K. Hujer, B. Decker, P. Rather, R.
Bonomo. Global Challenge of Multidrug-Resistant Acinetobacter baumannii. Antimicrobial agents and chemotherapy; 2007:3471–3484.
23. Lenie Dijkshoorn, A. Nemec, H. Seifert. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nature Reviews Microbiology 2007; 5: 939-951.
24. SHEA Guideline for Preventing Nosocomial Transmission of Multidrug Resistant Strains of Staphylococcus aureus and Enterococcus. Chicago journals 2003; 24(5): 362-86.
25. M. Aires de Sousa, Frequent Recovery of a Single Clonal Type of Multidrug-Resistant Staphylococcus aureus from Patients in Two Hospitals in Taiwan and China; J. Clin. Microbiol. 2003; 41: 1 159-163
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 816
ISSN 2229-5518
26. Richard H. Emerging Options for Treatment of Invasive, Multidrug-Resistant Staphylococcus aureus Infections. The Journal of Human Pharmacology and Drug Therapy 2007 ;
27: 227–249
27. J.M. Boyce. Outbreak of multidrug-resistant Enterococcus faecium with transferable vanB class vancomycin resistant. Journal of Clinical Microbiology 1994; 32 :1148-1153
28. BE Murray. Diversity among multidrug-resistant enterococci.
Emerging infectious diseases 1998; 4(1):37-47.
29. GD Bostic, Perri MB, Thal LA, Zervos MJ. Comparative in vitro and bacterial activity of oxazolidinone antibiotics against multidrug resistant enterococci. Diagnostic Microbiology and Infectious Disease 1998; 30: 109–112.
30. Sebaihia M, W ren BW , Mullany P, Fairweather NF, Minton N, Stabler R et.al. The multidrug resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nature genetics 2006; 38:779-786.
31. Keiko Hasegawa, N. Chiba, R. Kobayashi, S. Murayama, S.
Iwata, K. Sunaawa et. al. Rapidly increasing prevalence of β-lactamase-non-producing ampicillin-resistant Haemophilus influenza type b in patients with meningitis. Journal of Antimicrobial Chemotherapy, 57:1077-1082.
32. Jing-Jou Yan. Outbreak of Infection with Multidrug-Resistant Klebsiella pneumoniae Carrying blaIMP-8 in a University Medical Center in Taiwan. J. Clin. Microbiol. 2001; 39: 4433-
4439
33. Sonia Ktari, G. Arlet, Minif B. Gautier V, Mahajoubi F, Ben Jmeaa M. Emergence of Multidrug-Resistant Klebsiella pneumoniae Isolates Producing VIM-4 Metallo-β-Lactamase, CTX-M-15 Extended-Spectrum β-Lactamase, and CMY-4
AmpC β-Lactamase in a Tunisian University Hospital. Antimicrob. Agents Chemother. 2006; 50 : 4198-4201
34. Elizabeth B. Hirsch. Detection and treatment options for
Klebsiella pneumonia carbapenemases (KPCs): an
35. Cynthia G. W hitney, Increasing Prevalence of Multidrug- Resistant Streptococcus pneumoniae in the United States. N Engl J Med 2000; 343:1917-1924
36. C. Catchpole, A. Fraise, and R. W ise .Multidrug-Resistant
Streptococcus pneumonia. Microbial Drug Resistance 1996;
2(4): 431-432
37. Pablo J. Bifani. Origin and Interstate Spread of New York City Multidrug-Resistant Mycobacterium tuberculosis Clone Family JAMA. 1996;275 (6):452-457
38. A. Rattan, A. Kalia, N. Ahmad. Multidrug-resistant Mycobacterium tuberculosis: molecular perspectives. Emerg Infect Dis. 1998; 4(2): 195–209
39. Consuelo Beck-Sagué, Dooley SW , Hutton MD, Otten J, Breeden A, CrawfordJT et. al. Hospital Outbreak of Multidrug-Resistant Mycobacterium tuberculosis Infections: Factors in Transmission to Staff and HIV-Infected Patients. JAMA 1992; 268(10):1280-6.
40. Janak Koirala. Multidrug-resistant Salmonella enterica. The
Lancet Infectious Diseases 2011; 11 (11): 808 – 809.
41. M. Kathleen Glynn, Bopp C, Dewitt W , Dabney P, Mohtar M, Anqulo FJ. Emergence of Multidrug-Resistant Salmonella enterica Serotype Typhimurium DT104 Infections in the United States N Engl J Med 1998; 338:1333-1339
42. Ashraf Khan, Nawaz MS, Khan SA, Cerniqlia CE. Detection of multidrug-resistant Salmonella typhimurium DT104 by multiplex polymerase chain reaction, FEMS microbiology letters 2000; 182: 355–360.
43. Bernard Rowe. Multidrug-Resistant Salmonella typhi: A W orldwide Epidemic. Clin Infect Dis. 1997 ; 24(1): S106-9.
44. Peter Reichmann, Koniq A, Linares J, Alcaide F, Tenover FC, McDougal L, et.al. A Global Gene Pool for High-Level Cephalosporin Resistance in Commensal Streptococcus Species and Streptococcus Pneumoniae. J Infect Dis. 1997;
176 (4): 1001-1012.
emerging cause of multidrug-resistant infection. J Antimicrob | 45. | Harold C. Neu, The crisis in antibiotic resistance. |
Chemother 2010; 65 (6): 1119-1125 | 46. | Jeffrey Moscow , General Mechanisms of Drug Resistance |
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 817
ISSN 2229-5518
47. Brian Spratt, Resistance to antibiotics mediated by target alteration.
48. Georgopapadakou. Penicillin-binding proteins and bacterial resistance to beta lactam,; 1993: 2045 – 2053
49. Jitendra Vashist. Analysis of penicillin-binding proteins (PBPs) in carbapenem resistant Acinetobacter baumannii; Indian J Med Res 2011; 133 : 332-338
50. Nafisa H. Georgopapadakou and Fung Y. Liu. Penicillin- Binding Proteins in Bacteria. Antimicrobial agent and chemotherapy 1980 : 148-157
51. Gholizadeh, Y. and Courvalin, P. New strategies combating bacterial infection. International Journal of Antimicrobial Agents 2000; 16:S11–S17.
52. Popham and Setlow. Cloning, nucleotide sequence and regulation of the Bacillus subtilis pbpF gene, which codes for a putative class A high molecular weight penicillin binding protein. Journal of bacteriology 1993 : 4870-4876
53. Hartman and Tomasz. Low-affinity Penicillin binding protein associated with beta lactam resistance in Staphylococcus aureus; J of bacteriology 1984 : 513-516
54. Tiemersma E.W ., Bronzwaer S.L., Lyytikainen O., Degener, J.E., Schrijnemakers, P., Bruinsma, N., et al. European Antimicrobial Resistance Surveillance System Participants Emerging Infectious Diseases 2004; 10 : 1627–1634.
55. Crowcroft N.S. and Catchpole M. British Medical Journal
2002; 325:1390–1391.
56. Verma S, Joshi S, Chitnis V, Hemwani N, Chitnis D. Growing problem of methicillin resistant staphylococci – Indian scenario. Indian J Med Sci. 2000; 54:535–540.
57. John W . Dale-Skinner and Boyan B. Bonev. Molecular Mechanisms of Antibiotic Resistance: The Need for Novel Antimicrobial Therapies; W iley online library.
58. B.R.Lyon and R.Skurray. Antimicrobial resistance of
Staphylococcus aureus: genetic basis Microbiol Rev.1987;
51: 88-134
IJSER © 2013 http://www.ijser.org
59. K.Ubukata , K.Morakami and A. Tomaz. Use of the crystal structure of Staphylococcus aureus isoleucyl-tRNA synthetase in antibiotic design. Antimicrob Agents Chemother. 1985; 27,851
60. Predari, S. C., M. Ligozzi, and R. Fontana. Genotypic identification of methicillin-resistant coagulase-negative staphylococci by polymerase chain reaction. Antimicrob. Agents Chemother. 1991; 35:2568-2573.
61. Ubukata, K., S. Nakagami, A. Nitta, A. Yamane, S.
Kawakami, M. Sugiura. Rapid detection of the mecA gene in methicillin-resistant staphylococci by enzymatic detection of polymerase chain reaction products. J. Clin. Microbiol. 1992
; 30:1728-1733.
62. Ubukata, K., R Nonoguchi, M. Matsuhashi, and M. Konno.
Expression and inducibility in Staphylococcus aureus of the mecA gene, which encodes a methicillin-resistant S. aureus- specific penicillin-binding protein. J. Bacteriol. 1989
171:2882- 2888
63. Kayser. FemA, a host-mediated factor essential for methicillin resistance in Staphylococcus aureus: molecular cloning and characterization. Mol. Gen. Genet. 1989;
219:263-269.
64. Madiraju, M. V. V. S., D. P. Brunner, and B. J. W ilkinson.
Effects of temperature, NaCl, and methicillin on penicillin- binding proteins, growth, peptidoglycan synthesis, and autolysis in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 1987; 31:1727-1733.
65. Ubukata, K., R Nonoguchi, M. Matsuhashi, and M. Konno.
Expression and inducibility in Staphylococcus aureus of the mecA gene, which encodes a methicillin-resistant S. aureus- specific penicillin-binding protein.. J. Bacteriol. 1989;
171:2882- 288.
66. Severin A, Tabei K, Tenover F, Chung M, Clarke N, Tomasz A. High level oxacillin and vancomycin resistance and altered cell wall composition in Staphylococcus aureus carrying the staphylococcal mecA and the enterococcal vanA gene complex. J Biol Chem. 2004;279: 3398–3407.
International Journal of Scientific & Engineering Research, Volume 4, Issue 9, September-2013 818
ISSN 2229-5518
67. Perichon B, Courvalin P. Heterologous expression of the enterococcal vanA operon in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother.
2004; 48:4281– 4285.
68. Patrice Courvalin. Vancomycin resistance in gram positive cocci. Clin Infect Dis. 2006; 42 (Supplement 1): S25-S34.
69. Reynolds PE. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis 1989; 8:943–50.
70. Arthur M, Reynolds P, Courvalin P. Glycopeptide resistance in enterococci. Trends Microbiol 1996; 4:401–7.
71. Depardieu F, Reynolds PE, Courvalin P.VanD-type vancomycin-resistant Enterococcus faecium 10/96A. Antimicrob Agents Chemother 2003; 47:7–18.
72. Arias CA, Courvalin P, Reynolds PE. VanC cluster of vancomycin- resistant Enterococcus gallinarum BM4174. Antimicrob Agents Chemother 2000; 44:1660–6.
73. Abadia Patino L, Courvalin P, Perichon B. VanE gene cluster of vancomycin-resistant Enterococcus faecalis BM4405. J Bacteriol 2002; 184:6457–64.
74. Depardieu F, Bonora MG, Reynolds PE, Courvalin P. The vanG glycopeptides resistance operon from Enterococcus faecalis revisited. Mol Microbiol 2003; 50:931–48.
75. Rodríguez CA, Vesga O; Vancomycin-resistant
Staphylococcus aureus; Biomedica. 2005 ;25(4):575-87.
76. DC Hooper. Mechanism of fluroquinolone resistance; Drug
Resist Updat. 1999, 2(1): 38-55.
77. J Ruiz. Mechanism of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection. Journal of antimicrobial chemotherapy 2003;
51:1109-1117.
78. H Nikaido. Molecular basis of bacterial outer membrane permeability. Microbiological reviews 1985:1-35.
79. Anne H. Delcour. Outer Membrane Permeability and
Antibiotic Resistance; Biochim Biophys Acta. 2009; 1794(5):
808–816.
80. Nikaido H. Molecular basis of bacterial outer membrane permeability revisited Microbiol Mol Biol Rev 2003; 67:593–
656.
81. Vaara M. Agents that increase the permeability of the outer membrane Microbiol Rev 1992; 56:395– 411.
82. Richard Lewis. New compound defeats drug resistant bacteria;; Brown University, 28Nov 2011, 401-863-2476
83. Francoise Van Bambeke et al. Antibiotic Efflux Pumps; Biochemical Pharmacology 2000; 60: 457–470.
84. George, A.M. and P.M. Jones. Perspectives on the structure-function of ABC transporters: The Switch and Constant Contact Models. Prog Biophys Mol Biol. 2012;
109(3):95-107.
85. Ishii Y, Ohno A, Taguchi H, Imajo S, Ishiguro M, Matsuzawa
H. Cloning and sequence of the gene encoding cefotaximehydrolyzing class A ß-lactamase isolated from Escherischia coli. Antimicrob Agents Chemother 1995;
39:226
IJSER © 2013 http://www.ijser.org