The research paper published by IJSER journal is about Electronic Properties of Group V Substituted Fullerene: DFT Approach 1
ISSN 2229-5518
Electronic Properties of Group V Substituted
Fullerene: DFT Approach
Mohamed M. El-Okr, Abd el baset H. Makky, Hanan Elhaes and Medhat A. Ibrahim
Abstract— C59-X (X= C, N, P, As and Sb) was studied by Density Functional Theory DFT at the B3LYP/3-21G** level. Group V elements have been selected to be substituted in the fullerene-C60 cage in order to show the effect of such substations upon structural and electronic properties of the studied molecules. It is found that elements of Group V doped C60 have an electronic structure similar to those of n-type semiconductor. The electronic structure of electron donor (Group V-doped) of the C60 molecules demonstrates the possibility of using these systems in electronic applications as many properties as HOMO–LUMO gaps can be controlled by the appropriate substitution. At the same level of theory the effect of water, methanol, heptanes and acetonitrile upon C60, was studied in terms of geomet ry, HOMO/LUMO energy and total dipole moment. No changes in the calculated parameters were regarded as a result of solvents.
—————————— ——————————
ince the discovery of fullerene molecules C60, in 1985[1-2], considerable effort has been devoted to understanding the properties of this new molecule. Doped fullerenes have
also attracted a great deal of interest due to their remarkable structural, electronic, optical, and magnetic properties [3-4]. The unique structural and electronic properties of fullerenes led to another type of doping, named substitutional doping [3-
4], where one or more carbon atoms of fullerene are substi- tuted by other atoms, as well as the endohedral doping and exohedral doping. Molecular nanoscale electronic devices have attracted a great deal of attention in recent years [5]. Car- bon fullerene is one of the most stable and a well known nano- scale molecular structure; C60 often acts as a semiconductor. Density functional theory, (DFT) [6] arises from two theorems, the proofs of which were given by Hohenberg and Kohn [7].The use of computational methods, such as Density Func- tional Theory, to calculate structures of large numbers of mo- lecules is well established [8]. There have been a number of theoretical studies for electronic as well as physical properties of C60. These studies were carried out by semi–empirical me- thods [9], [11], as well as by ab initio and DFT calculations [12], [14]. Recently C60 continue to be the hottest research topic according to its emerging applications.
C60 was used for the growth of diamond by applying spark plasma sintering [15]. It is used efficiently for photo addition of the amino acid glycine as glycine methyl-ester to produce fulle- ropyrrolidine for many biological applications [16]. It is
————————————————
![]() Prof. Mohamed M. El-Okr is a Prof. of Solid State Physics, Physics
Prof. Mohamed M. El-Okr is a Prof. of Solid State Physics, Physics
Department Faculty of Science Al-Azhar University, Cairo, Egypt.
![]() Abdel-Baset H. Mekky* is an Instructor of Physics in Buraydah Colleges,
Abdel-Baset H. Mekky* is an Instructor of Physics in Buraydah Colleges,
Al-Qassim, Kingdom of Saudi Arabia. e-mail: hofny_a@mail.com
* Corresponding author
![]() Hanan Elhaes is an Associate Professor in the Physics Department, Faculty of Women for Arts, Science, and Education, Ain Shams University, Cairo, Egypt.
Hanan Elhaes is an Associate Professor in the Physics Department, Faculty of Women for Arts, Science, and Education, Ain Shams University, Cairo, Egypt.
![]() Prof. Medhat A. Ibrahim is the head of the Spectroscopy Department,
Prof. Medhat A. Ibrahim is the head of the Spectroscopy Department,
National Research Centre, Dokki, Cairo, Egypt.
also useful tool for fabrication of efficiently stable bulk hetero- junction solar cells [17].
Also it plays an important role as controlling the perfor- mance of polymer solar cells [18]. Finite element method (FEM) was used for vibrational and dynamic analysis of C60 and C30 [19].
Based upon above considerations DFT method at the B3LYP/3-21G* level was used to study C59X where X=C, N, P, As and Sb respectively. Geometry parameters, energy gap and total dipole moment were calculated at the same level of theory. The effect of solvents (water, methanol, heptanes and acetonitrile) upon C60 was studied in terms of total dipole moment and energy gap.
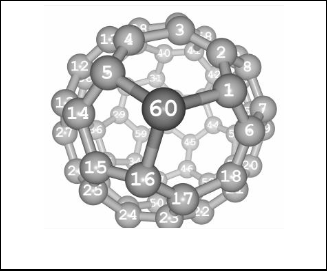
The C60 cage was adopted to provide an initial geometry to generate the heterofullerenes with only one carbon atom dis- placed by some elements from V Group. Accordingly a model of C59X was constructed as seen in figure 1 whereas X is subs- tituted with C, N, P, As, and Sb respectively. All geometries were fully optimized using the Gaussian 98 suite of programs on a personal computer [20]. Becke’s three-parameter hybrid exchange functional and the Lee-Yang-Parr correlation func- tional (B3LYP) [21], [23] was utilized with the 3-21G** basis set to optimize the studied structures. Solvent effects upon C60 were calculated at same level of theory by performing calcula- tions on the optimized structures using the CPCM (conducting polarized continuum model) [24], [25] method. The studied solvents are water, methanol, heptanes and acetonitrile respec- tively.
Fig. 1 presents the optimized geometry of C59X structure. The structure indicate that the X-atom (no 60) is substituted with C, N, P, As, and Sb respectively.
IJSER © 2012
The research paper published by IJSER journal is about Electronic Properties of Group V Substituted Fullerene: DFT Approach 2
ISSN 2229-5518
The minimal and maximal values of bond lengths are 1.385
Å and 1.464 Å respectively in case of C60. While the bond an-
gle found to be 108°(the angle between two adjacent single
C─C bonds) and 120° (the angle between a double C═C bond
and an adjacent single C─C bond).
Fig. 1. The Optimized structure of C59X, whereas the dopant is replaced with carbon atom number 60.
These substituents have selected in order to show the effect of doping on the structural and electronic properties of the fullerene molecules. The optimized bond lengths and bond angles for C60, C59N, C59P, C59As and C59Sb are listed in table 1, which obtained from DFT level calculations at B3LYP
/ 3-21g**. As indicated in the table 1 fullerene molecule has two different equilibrium bond lengths and two kinds of bond angles [26], [27].
TABLE 1
CALCULATED BOND LENGTHS AND BOND ANGLES AT
![]()
![]()
![]()
![]()
DFT-B3LYP/3-21G** LEVEL FOR C60 AND SPECIFIC FULLERENE DERIVATIVES.
Molecules | C60 | C59-N | C59-P | C59-As | C59-Sb |
Bond lengths (Å) | |||||
C C (min)a) | 1.385 | 1.383 | 1.382 | 1.38 | 1.383 |
C C (max)b) | 1.464 | 1.468 | 1.495 | 1.501 | 1.513 |
C X (min)c) | -------- | 1.359 | 1.796 | 1.935 | 2.131 |
C X (max)d) | -------- | 1.434 | 1.823 | 1.959 | 2.161 |
Bond angles (º) | |||||
C C C(min)e) | 108 | 107.3 | 107 | 106.7 | 106.3 |
C C C(max)f) | 120 | 121.5 | 123.5 | 124.2 | 125.2 |
C X C(min)g) | -------- | 107.6 | 88.5 | 83.3 | 121.1 |
C X C(max)h) | -------- | 121.4 | 123 | 122.9 | 123.1 |
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Note: In the Table X(dopant)= N, P ,As, and Sb
a) Minimal value of bond lengths between carbon atoms b) Maximal value of bond lengths between carbon atoms c) Minimal value of bond lengths between a dopant and
its nearest-neighbor carbon atoms
d) Maximal value of bond lengths between a dopant and its nearest-neighbor carbon atoms
e) Minimal value of bond angles between carbon atoms f) Maximal value of bond angles between carbon atoms g) Minimal value of bond angles between a dopant and
its nearest-neighbor carbon atoms
h) Maximal value of bond angles between a dopant and its nearest-neighbor carbon atoms
![]()
The optimized geometries for C59N, C59P, C59As and C59Sb clusters still keep the cage structures, but the regular pentagonal and hexagonal rings in C60 are now distorted in C59N, C59P, C59As and C59Sb. The various calculated C X bonds are in the range of 1.359–2.161Å. The bond lengths in- creased significantly on going from N toward Sb. As shown in figure 1, it can be expected, bond lengths increase down the group. This means that the stability for the group is decreased by substitution with heavier elements.
Another indication for the geometrical changes according to substations is indicated in terms the surrounding substi- tuted element. The substituted atoms (carbon atoms no 60) is surrounded with three carbon atoms make three bond lengths. The effect of substitutions will be discussed in terms of these three bond lengths. As shown in the table 2, the structure of fullerene, D60 - C1, D60 – C5 and D60 – C16 stand for three groups of non-equivalent carbon sites, there are two types of bond lengths (one pentagons and two hexagons), respectively. The bond lengths (D60 - C1, D60 – C5 and D60 – C16) have been selected to show the effect of substitutions upon the structure of C60. As indicated in the tables, the molecular point group is not changed and remains corresponding to "C1 "point group. Furthermore, except C59-N, all the studied structures have shown an enlargement in their bond lengths as compared with that of C60.
TABLE 2
CALCULATED BOND DISTANCE IN ANGSTROM AND MOLECULAR POINT GROUP FOR C60 AS W ELL AS DOPED C60, W HICH CALCULATED AT B3LYP/3-21G**.
Note: In the Table D = C, B, Al, Ga and In respectively.
The highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) energy separation serves as a simple measure of chemical stability [28-29]. This difference is known as the energy gap. Table 3 depicts the character of the HOMO/LUMO energy as (eV) at B3LYP/3-21G** level of C60 and specific fullerene derivatives.
Substitution of "dopant X" = N, P, As, and Sb, atoms on the fullerene C60 (C59X) affects the HOMO and LUMO energies. The calculated (HOMO–LUMO) energy gap of all these C59X clusters are reduced compared with the energy gap of C60. This means that in any excitation process C59-N, C59-P, C59-
IJSER © 2012
The research paper published by IJSER journal is about Electronic Properties of Group V Substituted Fullerene: DFT Approach 3
ISSN 2229-5518
As and C59-Sb molecules need more (ca. 1.14, 0.74, 0.69 and
0.086 eV, respectively) energy than that C60.
TABLE 3
CALCULATED HOMO/LUMO ENERGY AS (EV) AT
DFT-B3LYP/3-21G** LEVEL FOR C60 AND SPECIFIC FULLERENE DERIVATIVES.
![]()
![]()
Molecules C60 C59-N C59-P C59-As C59-Sb
Energy gap 2.89 1.14 0.74 0.69 0.086
![]()
This means that in any excitation process C59-N, C59-P, C59-As and C59-Sb molecules need more (ca. 1.14, 0.74, 0.69 and 0.086 eV, respectively) energy than that C60. It suggests that electrons in all C59X clusters are easier to excite from the HOMO to the LUMO than in the case of C60. One might expect to have broad range of new semiconductors because various gaps are formed with respect to the substituents and the range of substitution.
Table 4 shows the calculated total dipole moment for
C59X. The calculated total dipole moment was 1.2932, 0.7235,
1.3686 and 2.7509 debye for C59-N, C59-P, C59-As and C59-Sb
respectively. One can see from the table that the total dipole
moment value of C59-N, C59-P, C59-As and C59-Sb is higher
than that of C60.
TABLE 4
CALCULATED DIPOLE MOMENT AS DEBYE AT DFT-B3LYP/3-21G**
LEVEL FOR C60 AND SPECIFIC FULLERENE DERIVATIVES.
Dipole moment Contribution as debye | C60 | C59-N | C59-P | C59-As | C59-Sb |
X | 0.0000 | -0.0788 | -0.7195 | 1.3683 | -2.7507 |
Y | 0.0000 | -0.0811 | -0.0757 | -0.0305 | 0.0298 |
Z | 0.0000 | -1.2882 | 0.0000 | 0.0000 | 0.0000 |
Total dipole moment | 0.0001 | 1.2932 | 0.7235 | 1.3686 | 2.7509 |
C60 was subjected to different solvents namely water, me- thanol, heptanes and acetonitrile upon C60, was studied in terms geometry, HOMO/LUMO energy and total dipole moment. Table5. presents the calculated bond lengths (Å) and bond angles (º) for C60, which described through the polariza- ble continuum model (CPCM) and calculated by B3LYP me- thods with 3-21G** basis set. Results indicated that no change is regarded as a result of solvents. At the same level of theory the HOMO/LUMO energy gap as (eV) are calculated in listed in table 6. The studied solvents show no effect and the energy gap remains the same.
TABLE 5
CALCULATED BOND LENGTHS (Å) AND BOND ANGLES (º)
FOR C60, W HICH DESCRIBED THROUGH THE POLARIZABL E CONTINUUM MODEL (CPCM) AND CALCULATED BY B3LYP METHODS W ITH 3-21G** BASIS SET.
![]()
Solvent | Geometry Optimization | Reference | |||||||
CCa | CCb | CCCc | CCCd | CCa | CCb | CCCc | CCCd | ||
Gas Phase | 1.385 | 1.464 | 108 | 120 | 1.39 e | 1.46e | 108f, g | 120f, g | |
Water | 1.389 | 1.461 | 108 | 120 | |||||
Methanol | 1.393 | 1.457 | 107.9 | 120.2 | |||||
Heptane | 1.389 | 1.461 | 108 | 120 | |||||
Acetonitrile | 1.389 | 1.461 | 108 | 120 |
a) Minimal value of bond length between carbon atoms; b) Maximal value of bond length between carbon atoms c) Minimal value of bond angle between carbon atoms; d) Maximal value of bond angle between carbon atoms
e) David et al., 1991;
f) Yee et al., 2003
g) Tomekia et al., 2005
TABLE 6
CALCULATED HOMO/LUMO ENERGY GAP AS (EV) FOR C60,
W HICH DESCRIBED THROUGH THE POLARIZABLE CONTINUUM MODEL (CPCM) AND CALCULATED BY B3LYP METHODS W ITH 3-21G** BASIS SET.
![]()
![]()
Solvent | Energy Gap (eV) | Reference (Ying et al., 1998) |
Gas phase | 3.01 | 2.2553 |
Water | 3.007970264 | |
Methanol | 3.007970264 | |
Heptane | 3.007970264 | |
Acetonitrile | 3.007970264 |
![]()
Finally Table 7 presents the calculated dipole moment as well as its distributions in X, Y and Z directions. C60 shows
0.0 dipole moment this values remain the same and indicates that solvents are not affecting the dipole moment of the C60. A slight change is noticed only for the case of methanol
0.0006 debye.
TABLE 7
CALCULATED DIPOLE MOMENT AS DEBYE AT DFT-B3LYP/3-21G**
LEVEL FOR C60 AND SPECIFIC FULLERENE DERIVATIVES.
IJSER © 2012
The research paper published by IJSER journal is about Electronic Properties of Group V Substituted Fullerene: DFT Approach 4
ISSN 2229-5518
![]()
In this work we have studied the structural and electronic properties of C60 and some of its doped derivatives with some elements of group V by performing the Density Functional methods at B3LYP/3-21G** level. Calculated results of C59-N, C59-P, C59-As and C59-Sb molecules are compared with those of C60 at the same level of theory. It is found that these doped derivatives of fullerene have some remarkable features, which are different from and can com- pete with C60. For the various calculated C X bonds, the bond lengths for the heaver atoms increased significantly. This means that, the stability of group V is decreased by substitu- tion with heavier elements. There are no changes in the symmetry of the doped fullerenes, while the bond lengths are changed. Dipole moments of the doped C60 with some elements of group V are increased. The HOMO-LUMO energy gaps indicate that these are indeed new family semi- conducting systems in terms of various gaps. In summaries, the stability for the group is decreased by substitution with heavier elements. There exists a possibility to produce the semiconductor components of C60-X with various band gaps. Finally solvents show no change in the geometrical, band gap and dipole moment values.
[1] H W Kroto, J R Heath, S C O'Brien, R F Curl and R E Smalley, ―C60: Buck-
minsterfullerene‖, Nature, vol. 318, no. 6042, pp. 162-163, 1985.
[2] W. A. Krätschmer, L. D. Lamb, K. Fostiropoulos, and D. R. Huffman, ―Solid
C60: a new form of carbon ‖, Nature (London), vol. 347, pp.354-358, 1990.
[3] M. S. Dresselhaus, G. Dresselhaus, and P. C. Eklund, Science of Fullerenes and
Carbon Nanotubes, Academic Press, New York, NY, San Diego, CA, 1996.
[4] R. H. Xie, J. Zhao, and Q. Rao, ―Doped carbon nanotubes, in Encyclopedia of Nanoscience and Nanotechnology, ‖ edited by H. S. Nalwa, American Scien- tific Publisher, California, 2003.
[5] A. Nitzan and M. A. Ratner, ―Electron transport in molecular wire junctions‖,
Science, vol. 300, pp.1384, 2003.
[6] R. G. Parr and W. Yang., Density Functional Theory of Atoms and Molecules, Oxford University Press, 1989.
[7] P. Hohenberg and W. Kohn., ― Inhomogeneous Electron Gas‖, Phys. Rev. B,
vol.136, pp.B864-B871, 1964.
[8] W. Rudolph, and C. Pye, ― Raman spectroscopic measurements and ab initio molecular orbital studies of cadmium(II) hydration in aqueous solution‖, Journal of Physical Chemistry B, vol. 102, pp. 3564–3573, 1998.
[9] M. Ibrahim, H. ElHaes, A. J. Hameed, A. F. Jalbout and Aned de Leon, ―Anal- ysis of C60 Doping with Gallium, Indium and Phosphorus Using Semiempir- ical Molecular Modelling ‖, J. Comput. Theor. Nanosci., Vol. 6(1), pp. 85–88,
2009.
[10] M. Ibrahim, H. El-Haes, A. J. Hameed and A. F. Jalbout, ―Structural and Elec- tronic Properties of C60X6 (X= F, Cl, Br and I). A Theoretical Study‖, J. Com- put. Theor. Nanosci., vol. 5 (11), pp. 2247-2251, 2008.
[11] H. Elhaes and A. Babaier, ―Studying the Electronic Properties of Fullerene
Alkali Dimers‖, J. Comput. Theor. Nanosci., vol. 8, pp. 1509-1512, 2011.
[12] A. Ceulemans and P. W. Fowler, ―The Jahn–Teller instability of fivefold dege-
nerate states in icosahedral molecules‖, J. Chem.Phys., vol. 93, pp. 1221–1234,
1990.
[13] N. Manini, A. Dal Corso, M. Fabrizio, and E. Tosatti, ― Electron–vibration
coupling constants in positively charged fullerene ‖, Philos.Mag., vol. B 81,
pp.793–812, 2001.
[14] H. Elhaes, A. Babaeer and M. Ibrahim, ―Effect of Metal Substitution on the Electronic Properties of Fullerene and Fulleropyrrolidine‖ J. Comput. Theor. Nanosci., vol. 7, pp. 536-541, 2010.
[15] F. Zhang, F. Ahmed, G. Holzhuter, and E. Burkel, ―Growth of diamond from
fullerene C60 by spark plasma sintering‖, Journal of Crystal Growth, vol. 340,
pp.1–5, 2012.
[16] R. Skanji, M. B. Messaouda, Y. Zhang, M. Abderrabba, H. Szwarc, and F.
Moussa, ―Sequential photo-addition of glycine methyl-ester to [60]fullerene
‖,Tetrahedron, vol. 68, pp. 2713-2718, 2012.
[17] P. Wanga, K. Yao, L. Chen, Y. Chen, F. Li, H. Wang, and S. Yu, ―Self- assembled mesogens modified fullerene for efficiently stable bulk heterojunc- tion solar cells‖, Solar Energy Materials & Solar Cells, vol. 97, pp. 34–42, 2012.
[18] F. Zhang, Z. Zhuo, J. Zhang, X. Wang, X. Xu, Z. Wang, Y. Xin, J. Wang, J.
Wang, W. Tang, Z. Xu, and Y. Wang, ―Influence ofPC60BM orPC70BM as electron acceptor on the performance of polymer solarcells‖, Solar Energy Materials & Solar Cells, vol. 97, pp. 71–77, 2012.
[19] J.H. Lee, B.S. Lee, F.T.K. Au, J. Zhang, and Y. Zeng, ―Vibrational and dynamic
analysis of C60 and C30 fullerenes using FEM‖, Computational Materials
Science, vol. 56, pp. 131–140, 2012.
[20] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R.
Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Bu- rant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Mo- rokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cios- lowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaro- mi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al–Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head–Gordon, E. S. Replogle, and J. A. Pople, Gaussian 98, (Revision 5.2), Gaussian, Inc., Pittsburgh PA, 1998.
[21] A. D. Becke, ―Density-functional thermochemistry. iii. the role of exact ex-
change‖, J. Chem. Phys., vol. 98, pp.5648–5652, 1993.
[22] C. Lee, W.Yang, R.G. Parr, ―Development of the Colle-Salvetti correlation- energy formula into a functional of the electron density,‖ Phys. Rev. B, vol. 37, pp. 785-789, 1988.
[23] B. Miehlich, A. Savin, H. Stoll, and H. Preuss, ―Results obtained with the
correlation energy density functionals of becke and Lee, Yang and Parr,‖
Chemical Physics Letters, vol. 157, no. 3, pp. 200–206, 1989.
[24] V. Barone and M. Cossi, "Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model," J. Phys. Chem. A, vol. 102, pp. 1995–2001, 1998.
[25] J. Tomasi, B. Mennucci, and R. Cammi, ―Quantum mechanical continuum
solvation models,‖ Chem. Rev., vol.105, pp. 2999-3093, 2005.
[26] K. Hedberg, L. Hedberg, D. S. Bethune, C. A. Brown, H. C. Dorn, R. D. John- son, and M. de Vries, ―Bond lengths in free molecules of buckminsterfulle- rene, C60, from gas–phase electron diffraction, ‖ Science, vol. 254, pp. 410–412,
1991.
[27] M. S. Dresselhaus, G. Dresselhaus, and P. C. Eklund, Science of Fullerenes and
Carbon Nanotubes , Academic, New York, 1996.
[28] B. A. Hess, L. J. Schaad, "Hückel molecular orbital π re- sonance energies. The benzenoid ydrocarbons", J. Am. Chem. Soc., vol. 93, pp. 2413–2416, 1971.
IJSER © 2012