
International Journal of Scientific & Engineering Research, Volume 4, Issue 10, October-2013 262
ISSN 2229-5518
OP Gupta, Sita Rani
Abstract—Research in molecular biology has generated a huge amount of data. Various bioinformatics techniques are applied for the storage, analysis and manipulation of molecular data in different applications. One of the important application areas of bioinformatics is sequence analysis. Sequence analysis is used to identify structure, functions and features of long biological sequences. Sequence alignment is one of the key activities performed by bio-informaticians under molecular sequence analysis to identify the similarities between protein sequences or nucleotides. Many algorithms are available for molecular sequence alignment depending upon the type of application. Among one of the widely used algorithms are BLAST (Basic Local Alignment Search Tool), which identifies the regions of local similarity between the query sequence and library sequences. Depending upon the matches between the sequences similarity score is calculated. High score represents better alignment means close association between molecular sequences.
In this paper a new model of distributed BLAST for Local Area Network is proposed. In proposed algorithm query sequence is distributed by master node dynamically. Before query execution no of available slave nodes will be checked by master node and query sequence will be divided into same no of segments. In this implementation database will be replicated on all the slave nodes. Different algorithms will be executed by master and slave nodes of a LAN.
Index Terms- Algorithms; Bioinformatics; BLAST; Distributed Enviorment; FASTA; Sequence Analysis; Sequence Allignment;
-------------------- --------------------
ioinformatics is multidisciplinary field. It is the application of information technology and statistics in biology [1], [2] and [3]. In bioinformatics computers are used to store, analyze, manipulate and distribute biological data such as protein sequences, DNA or RNA [4] and [5]. Various application areas of bioinformatics include sequence analysis, genome annotation, analysis of gene expressions, comparative genomics, protein structure prediction etc [6]. Among these areas sequence analysis acts as fundamental step in many research applications. And key activity performed in analysis is sequence alignment which contributes to uncover genetic
evolutions of various organisms.
————————————————
• OP Gupta is currently working as Deputy Director of School of Electrical Engg. and Information Technology at Punjab Agricultural University, Punjab , INDIA , PH- 91-9872717300. E-mail: opgupta@pau.edu
• Sita Rani is currently pursuing Doctrate degree program in Computer
Science & Engg. from Punjab Technical University,Punjab, INDIA , PH-
91-9814216733. E-mail: sitasaini80@yahoo.in
In bioinformatics, comparison among the molecular sequences to identify regions of similarity is called sequence alignment. It is a key step in data processing in many bioinformatics applications. Its basic purpose is to reveal functional, structural or evolutionary relationships in different sequences. In sequence alignment regions in different sequences with maximum match are identified with some algorithms. Output of the algorithm is analyzed to judge the relationship between the biological properties of the compared sequences. There are two different well known techniques for sequence alignment i.e. Global Alignment and Local Alignment as shown in Fig 1. In Global Alignment every element of query sequence is aligned with compared sequence/sequences. This technique is preferred when sequences are almost of same length and similar. But when the sequences to be aligned are of different lengths and are dis-similar but are expected to contain some regions of similarity, Local Alignment is used. Sequence alignment can further be categorized as pair wise or multiple. Pair wise Sequence Alignment is used to identify similarities between two sequences at a time. In Multiple Sequence Alignment more than two sequences can be considered at same time for best possible alignment.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 10, October-2013 263
ISSN 2229-5518
Fig 1. Global and Local Sequence Alignment
Different bioinformatics algorithms are available for different types of sequence alignment. A well known global alignment method is Needleman-Wunsch algorithm and famous local alignment methods are Smith-Waterman algorithm and BLAST (Basic Local Alignment Search Tool). Depending upon type of application one or another algorithm is used for sequence analysis [7], [8] and [9].
BLAST is the most commonly used algorithm to execute queries on biological databases. When a query sequence is given, the motive is to find similar sequences from the available databases. Similar sequences can also be searched by calculating the pair wise score of the alignments between the query sequence and the database sequences, as it is done in the Smith – Waterman algorithm. But in this computational cost is very high. BLAST uses some heuristic methods to reduce the running time with little sacrifice in accuracy [10].
BLAST is used to identify the regions of local similarity between query sequence and database sequences by considering some threshold value. For BLAST algorithm, input is given in FASTA format. FASTA format is represented as: >gi|5524211|gb|AAD44166.1| cytochrome b
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYV LPWGQMSFWGATVITNLFSAIPYIGTNLVEWIWGGFSVDK ATLNRFFAFHFILPFTMVALAGVHLTFLHETGSNNPLGLTS DSDKIPFHPYYTIKDFLGLLILILLLLLLALLSPDMLGDPDNH MPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSI VILGLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWI GSQPVEYPYTIIGQMASILYFSIILAFLPIAGXIENY
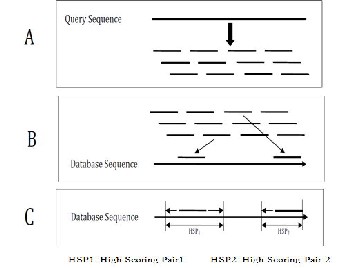
Where “>” sign acts as sequence identifier and remaining text gives the description. So both the query sequence and database sequence need to be converted into FASTA format before applying the algorithm. BLAST’s final result consists of a series of local alignments, ordered by the similarity score achieved in the extension process. It presents the complete sequences matched, score, size of the alignment and also a value based on the probability that the alignment has been found by chance, the e-value. The e-value depends on the query and database composition, and some of the alignments with higher e-values are discarded from the result. A simplified implementation of BLAST is shown in Fig 2. [12]
Fig 2. Simplified Implementation of BLAST
Because of the variety of the biological sequences to be worked upon, different variations of BLAST i.e. blastn, blastp, PSI-BLAST, blastx and many other are available [11].
Although BLAST is time efficient as compare to other dynamic programming algorithms but as the size of the databases is very huge, query execution time is still a limiting factor. To reduce the time associated with query execution, many efforts are being put in the direction of parallel sequence alignment with BLAST [12]. Various parallel implementations of BLAST are achieved either by query segmentation or by database segmentation or both. BeoBLAST is a parallelized implementation of BLAST developed to run on Beowulf clusters. Beowulf clusters do not behave like network of independent nodes but as acts as a single system. These are scalable clusters and run on LINUX or UNIX. In Beowulf clusters BLAST is parallelized by using query segmentation technique. In this implementation speed-up achieved is not super linear [13]. MpiBLAST is another parallel and most popular version of BLAST. In mpiBLAST both query and database segmentation is being used. It is designed to execute on distributed systems which are already designed. MpiBLAST has become successful in achieving super linear speed up [14]. Soap-HT-BLAST is a web based distributed implementation of BLAST. Perl and Apache Web Server are required to be installed on the system to use this implementation. As Soap-Ht-BLAST is a web based application so nodes distributed across World Wide Web can be used as workers. But the major drawback is the latency because of remotely distributed nodes and achieved speedup is not super linear [15].
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 10, October-2013 264
ISSN 2229-5518
The basic purpose of the distributed computing environment is to perform distributed computations. The frame work for this implementation is designed to be capable of keeping data about slave nodes, identifying idle slave nodes and distributing data & executing tasks among the available nodes. Every time before execution of a query master node determines idle nodes ready to be assigned next tasks. Accordingly query sequence is divided into different segment and distributed for execution. Different algorithms are executed by master and slave nodes. In this model of BLAST only query segmentation is applied and same database is replicated on all the nodes.
Steps:
1. Master node will identify all idle nodes ready to be
assigned next task for execution.
2. Query sequence will be divided into number of segments equal to available worker nodes.
3. Once again node availability will be confirmed.
4. Generated query segments will be distributed among
worker nodes.
5. Master node will assure that during execution all the required data is available with slaves.
6. Results will be collected from various nodes and compiled.
Steps:
1. Slave will establish a connection with master and
transfer its status.
2. Receive the query segment from master.
3. Execute the query.
4. If any data required is not available, intimate to master.
5. To send the results to master node.
The model is tested for two types of environments. In Type A environment BLASTN is implemented on four machines each running Windows Server 2003. Each machine was with two four core processors and 16 GB RAM. In Type B environment, there were six systems with Windows Server 2008 operating system and two four core processors with 6 GB RAM. Both the time same database is used. Database was having around
150,000 sequences.
In the proposed distributed implementation of BLAST query segmentation is applied for parallelization. Query segmentation is done dynamically as per availability of free worker nodes in LAN. Results are shown by implanting new model in two different environments. Time of distributed implementation is compared with linear implementation for both the environments.
In current work only BLAST is implemented on distributed environments. In future, this research work can be extended to develop general distributed bioinformatics applications to enhance the process of sequence analysis. The model can be implemented on more number of machines. The model can further be enhanced to work on heterogeneous nodes.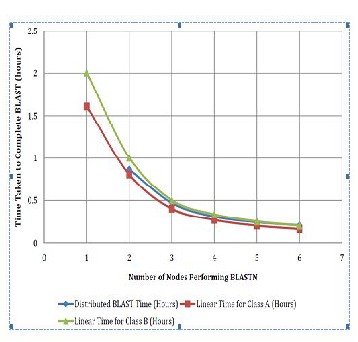
Fig 3. Model Implementation Results
[1] W. Feng, “Massively Parallel Sequence Alignment: When Computational Biology Becomes I/O Biology”, Populations, Communities, Ecosystems and Evolutionary Dynamics: Genomics and Metagenomics, vol. 10, no. 4, pp. 19-22, 2009.
[2] P.L. Mcmahon, “ Accelrating Genome Sequence Alignment using High Performance Reconfigurable Computer”, Ph.D Dessertation, University of Cape Town, 2008.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 4, Issue 10, October-2013 265
ISSN 2229-5518
[3] D. Diaz, F.J. Esteban, p. Hernandez, J.A. Cabbalero, G. Dorado and S.
Galvez, “ Parallelizing and optimizing a bioinformatics sequence alignmnet for many-core architecture”, Parallel Computing, vol. 11, pp. 244-259, 2011.
[4] A. Wirawan, C.K. Kwoh, N. Hieu and B. Schmidt, “ CBESW: Sequence Alignmnet on Playstation 3”, BMC Bioinformatics, pp. 1-10,
2008.
[5] G.J. Barton, “Sequence alignment for Molecular Replacement”,
Biological Crystallography, vol. 9, no. 2, pp. 25-32, 2008.
[6] S. Chen, C. Lin and S.Chen, “ Multiple DNA Sequence Alignment Based on Genetic Algorithms and Divide-and-Conquer Techniques”, International Journal of Applied science and Engineering, vol. 3, no. 2, pp.
89-100, 2005.
[7] L.V. Vinh, T.V. Lang, N.T. Du and V.H. Chau, “ Multiple sequence Alignment using Grid Computing using Cache Technique”, International Journal of Computer Science and Telecommunication, vol. 3, no. 7, pp. 46-51, 2012.
[8] H. Mathkour and M. Ahmed , “ A Comprehensive Survey on Genome sequence Analysis”, IEEE International Conference on Bioinformatics and Biomedical Technology, pp. 14-18, 2010.
[9] Y. Chen, S. Yu and M. Leng, “Paralleln sequence Alignment Algorithm for Clustering System”, International federation for Information Processing, pp. 311-321, 2006.
[10] S.F. Altschul, G. Warren, W. Miller, W.M. Eugene and D.J. Lipman, “ Basic Local Alignment search Tool”, Journal of Molecular Biology, vol.
215, no. 3, pp. 403-410, 1990.
[11] C. Noteredame, “Recent Evolutions of Multiple Sequence Alignment
Algorithms”, PLOS Computational Biology, vol. 3, no. 8, pp. 1405-1408,
2007.
[12] E. J. Ree, “ Parallelization Methods for the Distribution of High- Throughput Bioinformatics Algorithms”, Ph. D. Dessertation, Texas Tech University, 2011.
[13] J.D. Grant, R.L. Dunbrack, F.J. Manion and M.S. Ochs, “ BeoBLAST: distributed BLAST and PSI-BLAST on a Beowulf Cluster”, Bioinformatics Applications Notice, pp. 765-766, 2002.
[14] A.E. Darling, C. Lucas W. Feng, “The Design , Implementation and Evaluation of mpiBLAST”, 4th International Conference on LINUX Clusters: The HPC Revolution 2003 in conjuction with ClusterWorld Conference and Expo, 2003.
[15] W. Jiren and Q. Mu, “ Soap-HT-BLAST: High Throughput BLAST on b Services”, Bioinformatics Applications Note, pp. 1863-1864, 2003.
IJSER © 2013 http://www.ijser.org
International Journal of Scientific & Engineering ResearcVolume 4, Issue 10, October-2013
ISSN 2229-5518
266
266